
To the Editor:
Bardet in 1920 and Biedl in 1922 described a congenital syndrome characterised by obesity, polydactyly, retinitis pigmentosa, mental retardation and gonadal hypoplasia. Other frequently associated abnormalities are high blood pressure and diabetes mellitus.1,2 Adult patients consult nephrologists due to high blood pressure, abnormal renal imaging tests or kidney failure, which is the greatest cause of morbidity and mortality.1
EPIDEMIOLOGY AND DIAGNOSIS
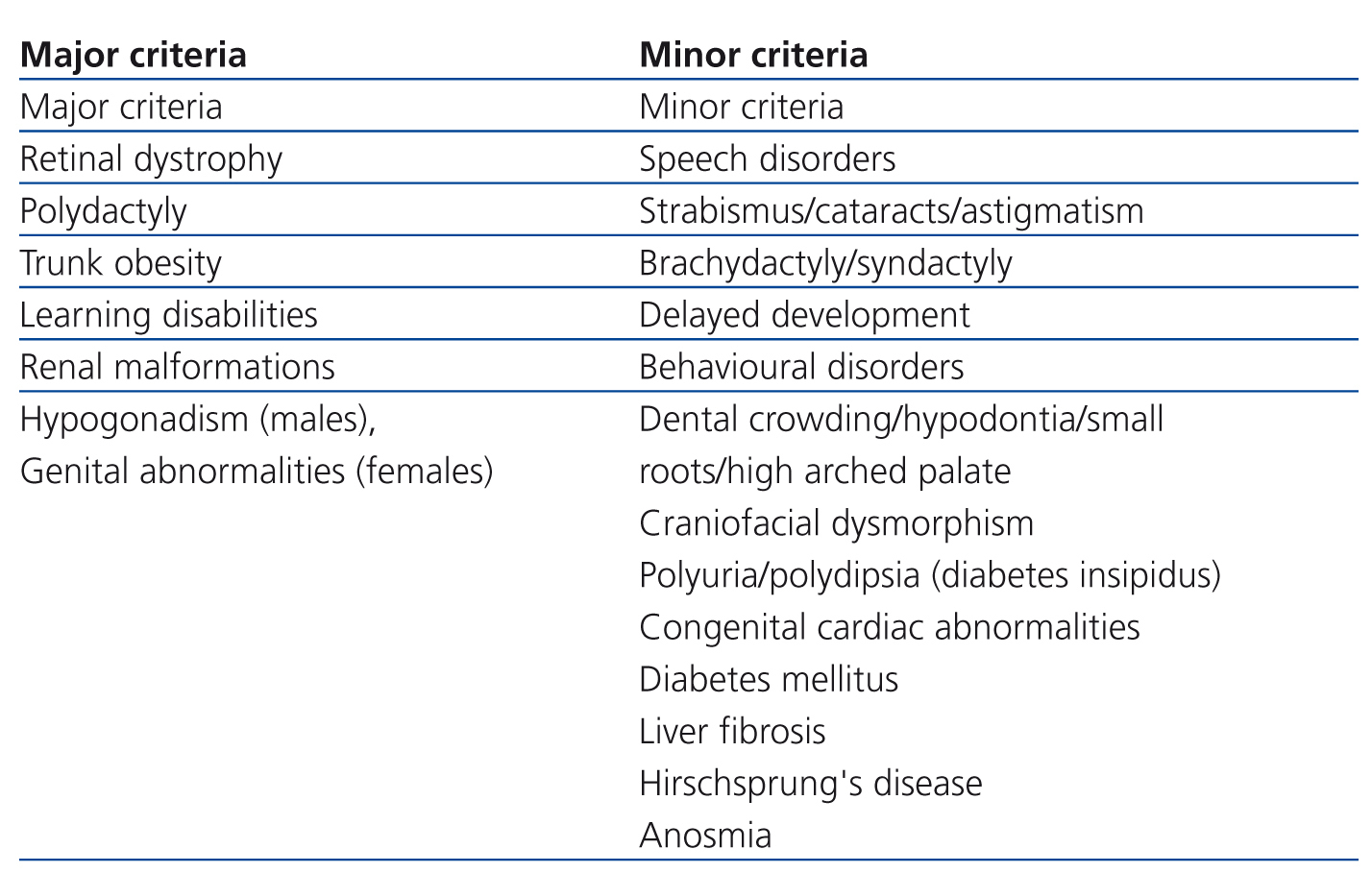
Incidence varies geographically between 1/160,000 in Switzerland and 1/13,500 amongst Bedouins of Kuwait, and is probably associated with endogamy.1 Diagnosis is mainly clinical. In 20011 Beales et al. suggested that the presence of three or four major criteria associated with two minor criteria may be diagnostic (Table 1), but diagnosis may be delayed due to slow emergence and clinical variability in disease expression. Clinical differential diagnoses of an obese child with psychomotor retardation and without polydactyly involve a wide range of possibilities, until visual impairment appears.3
GENETICS OF THE BARDET-BIEDL SYNDROME
There are at least 16 genes related to the Bardet-Biedl syndrome (BBS), including BBS1, BBS2, ARL6/BBS3 and SDCCAG8/BBS16. In the autosomal recessive mode of inheritance, parents are obligate heterozygotes and asymptomatic, while siblings will have a 25% chance of being healthy, 50% will be heterozygous and 25% will be affected by the disease. Triallelic inheritance implies the presence of at least three mutations for the phenotype to manifest. It is currently unknown to what extent multiple allelism contributes to the phenotype.1,4
RENAL MALFORMATIONS AND ABNORMAL RENAL FUNCTION
Renal involvement is observed in up to 40% of cases. Renal dysplasia is characterized by abnormalities in the parenchyma and nephronophthisis. Lobulations are found by ultrasound and nuclear magnetic resonance. Symptoms include anaemia, polyuria and polydipsia in childhood. Glomerular disease is less common and includes focal segmental glomerulosclerosis and glomerular basement membrane detachment, although renal biopsy is rarely performed.5
TREATMENT
There is no treatment for loss of vision, but special education and future training for blindness can be provided. Obesity should be managed by diet and exercise. Dyslipidemia and high blood pressure are treated as usual and specialized monitoring will be given in accordance to the degree of renal failure. Genetic counselling should be offered. 1,6
CASE STUDY
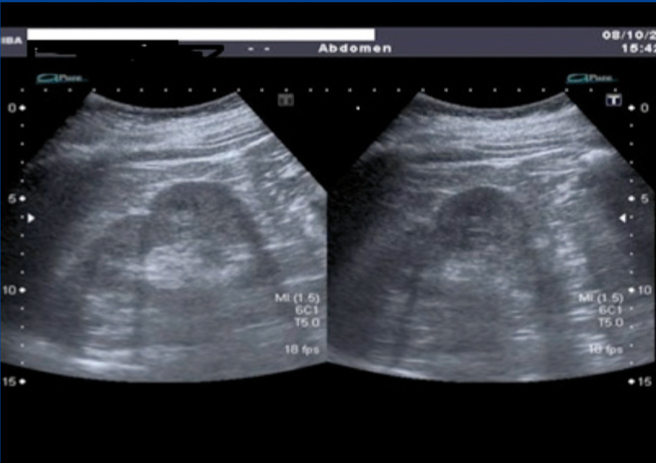
A 50-year-old female without allergies; underwent surgery due to polydactyly at two years of age. Retinitis pigmentosa was diagnosed more than 25 years ago. Active smoker (20 packs/year), with chronic obstructive pulmonary disease (chronic bronchitis). She denied blindness in first and second degree family members. Physical examination: weight: 85.9kg, height 1.65m, body mass index: 31.55kg/m2. Blood test without anaemia, dyslipidemia nor increased serum creatinine. Genetic study in the Fundación Jiménez Díaz in Madrid in May 2010: carrier of a mutation in gene BBS1, which is responsible for her illness. Abdominal ultrasound (Figure 1): fatty liver, pancreas not assessable, spleen homogeneous. Right kidney with small lobulations and a 3.8cm sinus cyst, left kidney with rounded cortical prominence, likely lobulation, but a renal tumour could not be ruled out, and therefore nuclear magnetic resonance was advised, but it was not performed because of claustrophoby.
BARDET-BIEDL SYNDROME AS A CILIOPATHY
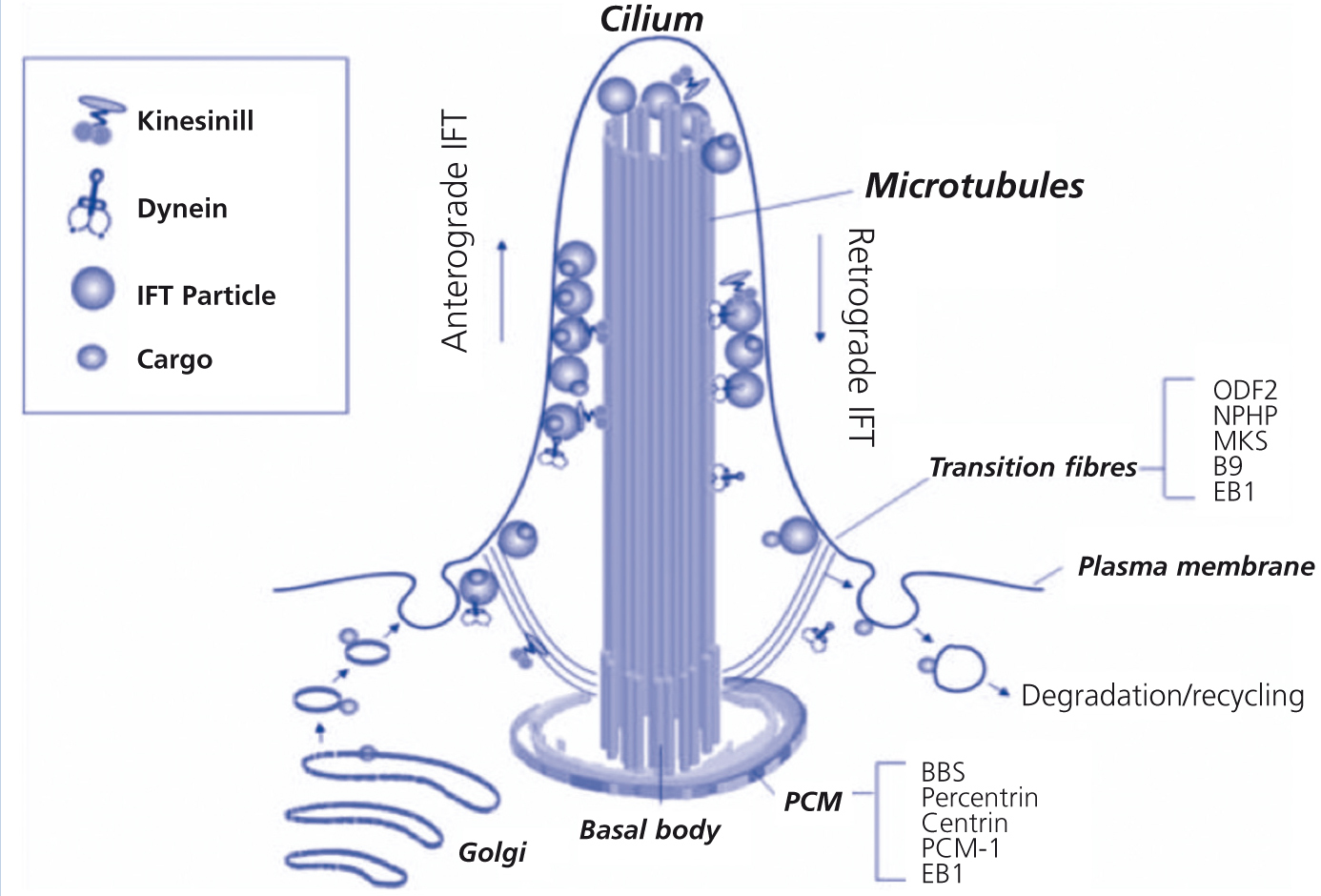
Cilia are present in almost all vertebrate cells. They are involved in embryonic development, polarity and homeostasis, sensory functions (hearing, sight, smell), transport functions, and cell division. Their assembly and maintenance depends on intraflagellar transport, in which basal body particles are carried along the microtubule structure to the tip. In 2003, Ansley et al.7 proposed that the molecular pathogenesis of BBS was due to ciliary involvement. Many proteins encoded by abnormal genes in BBS are located in the centrosome and basal body. Glomerular and tubular cells produce a single cilium with a 9+0 structure that projects into the tubular lumen and functions as a mechanoreceptor or chemoreceptor. Other ciliopathies include Kartagener’s syndrome, AD polycystic disease and nephronophthisis8 (Figure 2).
CONCLUSIONS
BBS is a very rare genetic disease. Although clinically it is well defined and characterized by the association of polydactyly, retinitis pigmentosa, psychomotor retardation, and obesity, genetically it is heterogeneous, and includes genes that encode for proteins essential for the functioning and structure of cilia, and as such, this syndrome is grouped within ciliopathies, pathogenic entities that have sparked renewed scientific interest. On reviewing the literature, we found that in Castile and Leon (Spain) 4 cases in total have been reported, corresponding to 2 families with 2 affected siblings each.9,10
Conflicts of interest
The authors declare that they have no conflicts of interest related to the contents of this article.

Figure 1. Abdominal ultrasound showing lobulations in both kidneys.

Figure 2. The cilium.

Table 1. Bardet-Biedl syndrome diagnostic criteria (Beales et al.).



