
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disorder. Its estimated prevalence is 1 per 800 individuals. ADPKD patients constitute 8% of the population on dialysis or kidney transplantation. The disease can be diagnosed using radiological or genetic procedures. Direct genetic diagnosis of the disease can now be performed in Spain; however, it is not an easy or cheap test. This is why every case should be considered individually to determine whether genetic testing is appropriate, and to determine which genetic test is most adequate. Genetic testing in ADPKD is of special interest for living donors and neonatal and sporadic cases. Genetic testing offers the chance of performing prenatal or pre-implantation testing of embryos in families with severe cases of the disease. Also, this will enable the disease to be treated, when specific treatment becomes available, in cases that would not be candidates for treatment without genetic confirmation.
La poliquistosis renal autosómica dominante (PQRAD) es la enfermedad renal hereditaria más frecuente. Su prevalencia estimada es de 1 cada 800 personas. Los pacientes con PQRAD constituyen un 8% aproximadamente de la población en diálisis o trasplante renal. El diagnóstico de la enfermedad es radiológico y/o genético. La posibilidad de realizar un diagnóstico genético directo de la PQRAD es actualmente una realidad en nuestro país, aunque por las características del gen PKD1 no es un análisis sencillo ni económico. Debe estudiarse cada caso de forma individualizada con el fin de determinar la idoneidad de realizar un estudio genético y determinar qué tipo de estudio es el adecuado. El diagnóstico genético es de especial interés para los donantes vivos, para casos neonatales y para casos esporádicos. El diagnóstico genético permite ofrecer diagnóstico prenatal o preimplantacional en familias con casos severos de la enfermedad y también permitirá tratar la enfermedad, cuando exista un tratamiento específico, en aquellos casos dudosos que sin confirmación genética no serían candidatos a tratamiento.
INTRODUCTION
Clinical and epidemiological background
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease. Its prevalence is estimated at 1 in 800 people. About 8% of patients on dialysis or undergoing kidney transplantation have ADPKD. It is characterised by the progressive development of kidney cysts that often lead to end-stage kidney disease (ESKD), usually in adulthood. Most patients usually have liver cysts, but only a minority develops massive polycystic liver disease. The presence of cerebral aneurysms in these patients is also rare (approximately 10%). A familial clustering of cerebral aneurysms has been described.1,2 Therefore, cranial magnetic resonance angiography is currently recommended when an affected family member has brain aneurysms or an incident compatible with ruptured aneurysms. There is considerable clinical variability both within and between families, with the former being much more common.3 The phenotypic spectrum of the disease ranges from severe prenatal cases to elderly patients with normal kidney function.
Genetic background
It is well known that the disease ADPKD has a genetic cause. It has genetic heterogeneity, i.e. more than one gene causes the disease. The genes responsible are: PKD1 (located on chromosome 16, at 16p13.3)4 and PKD2 (located on chromosome 4, at 4p21).5 Mutations in the PKD1 gene are responsible for 85% of ADPKD cases, while mutations in the PKD2 gene give rise to the remaining 15%.6 The disease is more severe in cases caused by PKD1 mutations. The mean age at onset of dialysis is 54.3 years for people with PKD1 and 74 for people with PKD2.7 For the same age, kidney size is significantly higher if the gene responsible is PKD1 than for PKD2, which fully justifies the different evolution of the disease according to the CRISP study.8 There is, however, considerable variability in the age of dialysis onset for each of the genes.9
PKD1 gene
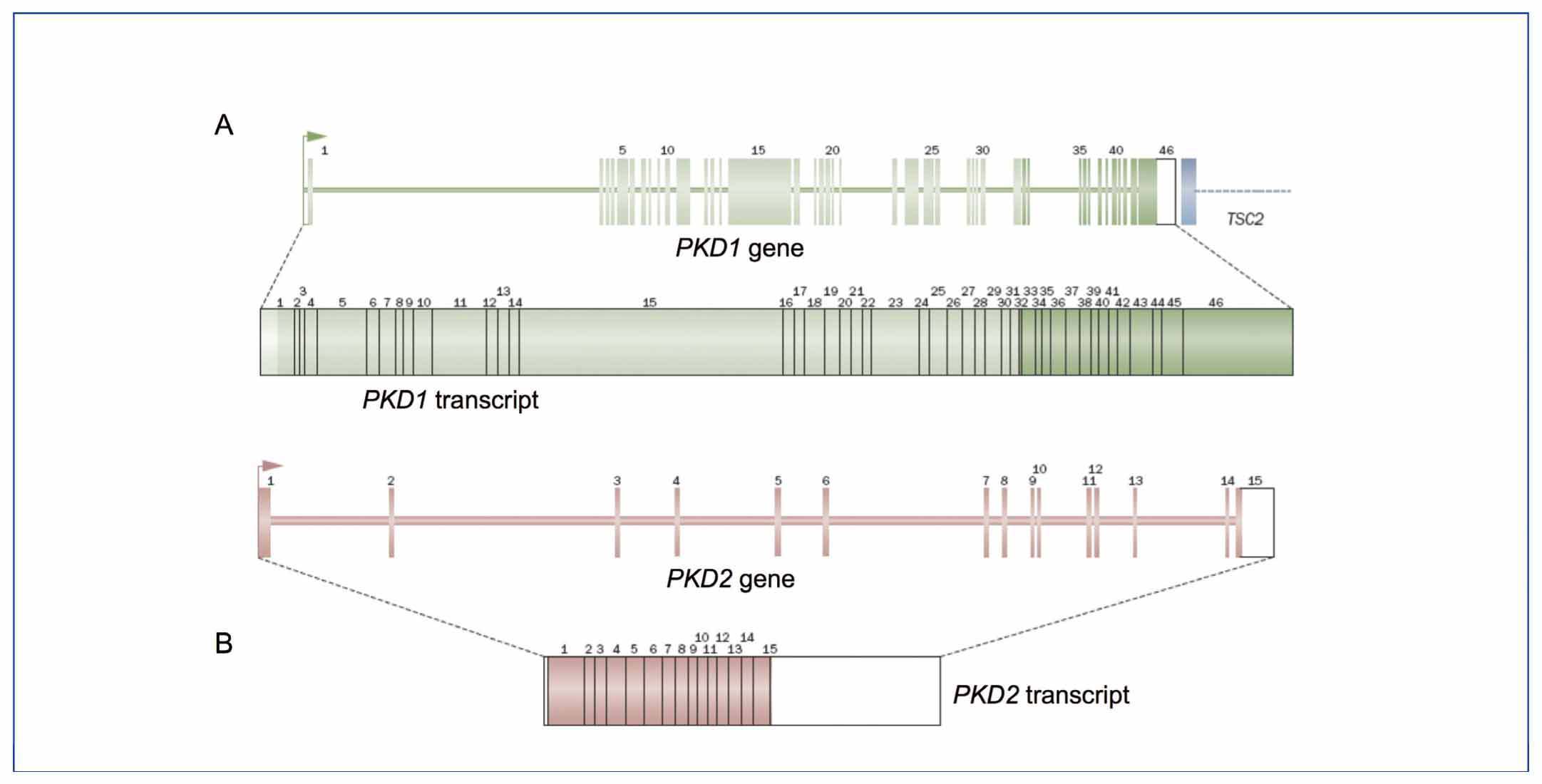
The PKD1 gene consists of 46 exons and encompasses a 54kb genomic region. It is transcribed into an mRNA of approximately 14kb with a 12,909bp coding sequence (Figure 1A).10,11 Analysis of this gene is very complex, because the region between exons 1 and 33 has undergone intrachromosomal duplication throughout human evolution. It now contains 6 pseudogenes (PKD1P1-P6), located between 13 and 16Mb proximal to the PKD1 gene. These pseudogenes are expressed but have stop codons preventing translation, so they are expected give rise to small non-functional proteins. By comparing the sequences of these pseudogenes with PKD1 pseudogenes, it can be seen that they contain mutations but they have a sequence identity of 98-99% in regions homologous to PKD1. The minor differences between these pseudogene sequences and PKD1 have been fundamental in designing strategies for the selective amplification of PKD1, thereby preventing the amplification of these pseudogenes.12-14
The PKD1 gene encodes an integral membrane protein called polycystin 1.
PKD2 gene
The PKD2 gene contains 15 exons and covers a genomic region of 68kb with a coding sequence of 2,904pb.5,15 This gene encodes the protein polycystin 2, which is a TRP calcium channel family (transient receptor potential). This is why it is also called TRPP2 (Figure 1B).
Both polycystins are located in the primary cilia, among other places. Therefore, ADPKD is considered a ciliopathy, like all cystic diseases of the kidney. It seems that the polycystins have a function related to the detection of flow,16 pressure17 and modulation of centrosome duplication, and/or cell cycle regulation.18,19 They have also been implicated in the preservation of planar polarity.20 All these mechanisms may be involved in cystogenesis, but the precise key mechanism is still unclear.
DIAGNOSIS OF AUTOSOMAL DOMINANT POLYCYSTIC KINDEY DISEASE
Since ADPKD is an autosomal dominant disease with high penetrance, the offspring of affected parents have a 50% chance of developing the disease. In addition, given its high penetrance, it would be highly unlikely to clinically skip a generation. Currently, the disease is usually ruled out or confirmed after ultrasound in a person with an affected relative. The diagnosis of the disease is at present radiological and/or genetic.
Radiological diagnosis
Ultrasound is the most widely used technique for diagnosing the disease in subjects at risk. The classic criteria for the diagnosis of PKD1 were described by Ravine et al. in 1993,21 and more recently, the ultrasound criteria for ADPKD when the gene causing the disease is unknown have been published.22
There is no family history of the disease in 10% of cases found.23,24 In these cases, the disease is diagnosed by chance, but it is often difficult to determine if it is really an ADPKD. The general population can progressively develop kidney cysts with age, so diagnosis is more complex for cases without a family history or in elderly adults. Despite the relative accuracy of ultrasound criteria, they are not very sensitive or specific in the young and in elderly individuals with a mild form of the disease.
CT and MRI are more expensive techniques than ultrasound but are more sensitive, with smaller cysts being detectable (2mm instead of 10mm with ultrasound). There are no radiologic criteria for diagnosis of ADPKD by CT or MRI.
The difficulty of diagnosing ADPKD via radiology in certain cases, such as the young or those with no family history, have necessitated the development of genetic diagnosis.
Genetic diagnosis
Genetic linkage analysis
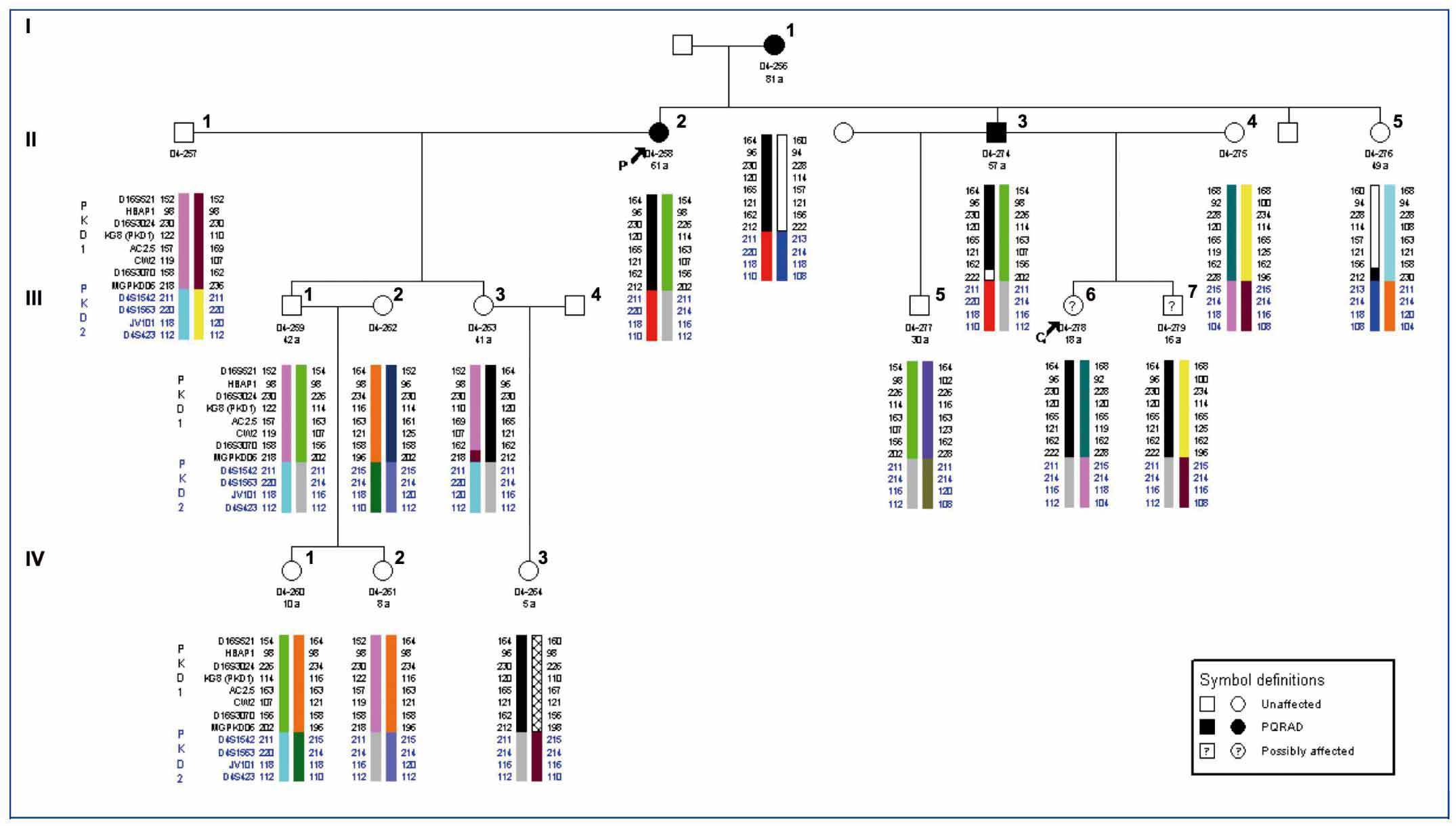
Locating the genes PKD1 and PKD2 allowed ADPKD to be diagnosed by genetic linkage analysis in the 1990s. This study is indirect and is based on the analysis of genetic markers located in the regions of the PKD1 and PKD2 genes (Figure 1). It can determine whether it is the PKD1 or PKD2 haplotype which segregates with the disease in a given family and, consequently, which gene is responsible for the illness in the family. Currently, there are databases available locating a minimum of 15 microsatellite markers useful for genetic linkage to PKD1 and 8 markers for PKD2. The major drawback of this type of diagnosis is that it can only be used in family cases. Also, due to the genetic heterogeneity of the disease, there must be several other affected and unaffected family members available for radiological study as well as the patient for an accurate ascertainment of the disease. It is also imperative that one member of the family is diagnosed with ADPKD with absolute certainty (this person is called the testing or index case). Only certain families are large enough for the study to confirm the linkage to one of the genes and discard the linkage to the other gene. In some cases, all affected family members share both the PKD1 and the PKD2 haplotype, and none of the unaffected family members carry these. Therefore, the study does not provide any information (as it cannot be discerned if the PKD1 or PKD2 is responsible for the disease, Figure 2). However, it is not only family size or how informative the markers may be that make this such a complicated diagnostic approach. There are also other phenomena, such as the presence of de novo mutations, mosaicism, the presence of hypomorphic alleles, etc. All this indicates that one must be very cautious with this type of diagnostic approach.
Mutation analysis
Although there are various possible approaches for the mutation study of PKD1 and PKD2 genes, exon sequencing is the most commonly used technique today. Many studies have shown the allelic heterogeneity of these genes, such that the same mutation is not found in more than 2% of families. This fact greatly complicates the search for mutations. Therefore, all exons in the PKD1 and PKD2 genes must be analysed systematically. Given the size and complexity of PKD1, this is particularly expensive. To analyse the PKD1 region homologous to pseudogenes, polymerase chain reaction (PCR) of the sequences where PKD1 differs from these pseudogenes has been designed. Thus, the genomic region that includes exons 1 to 33 is amplified in 5 long PCR, with these initial products then used as a template in the subsequent amplification of these 33 exons.13 The remaining 13 PKD1 and 15 PKD2 exons can then be amplified conventionally using the patient’s genomic DNA. Once the 46 exons in PKD1 and/or the 15 in PKD2 are sequenced, the potential pathogenicity of the different sequence variants identified must be correctly assessed.
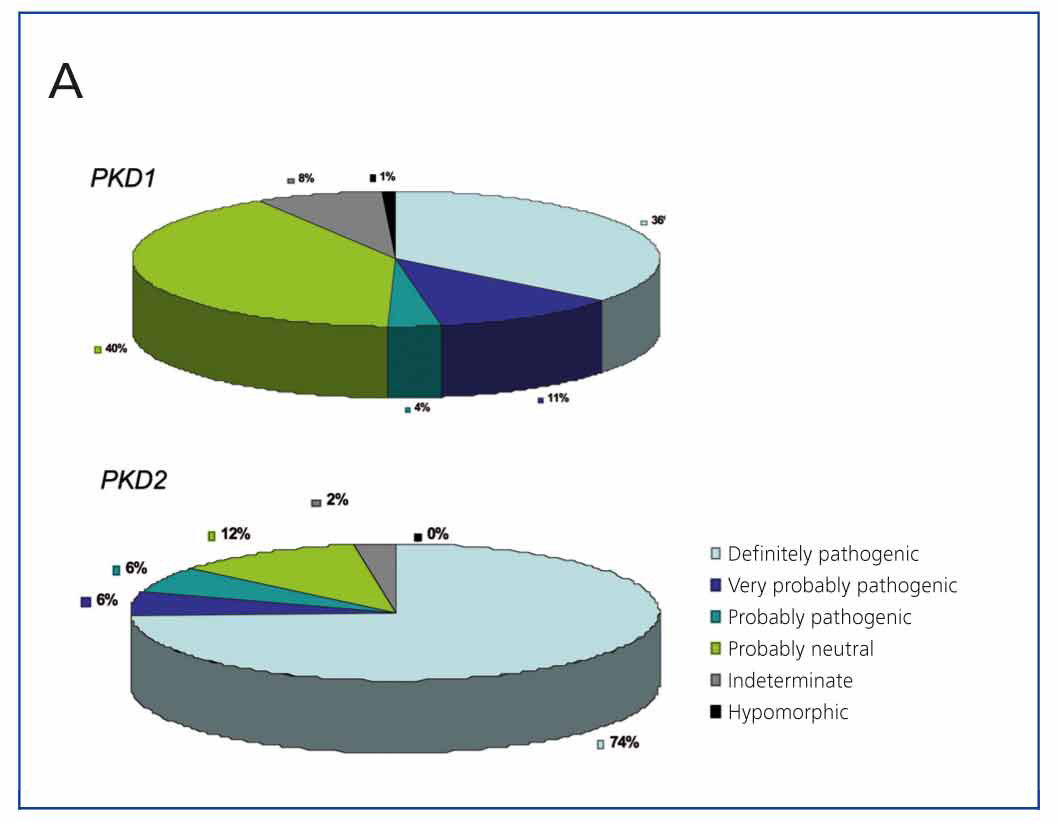
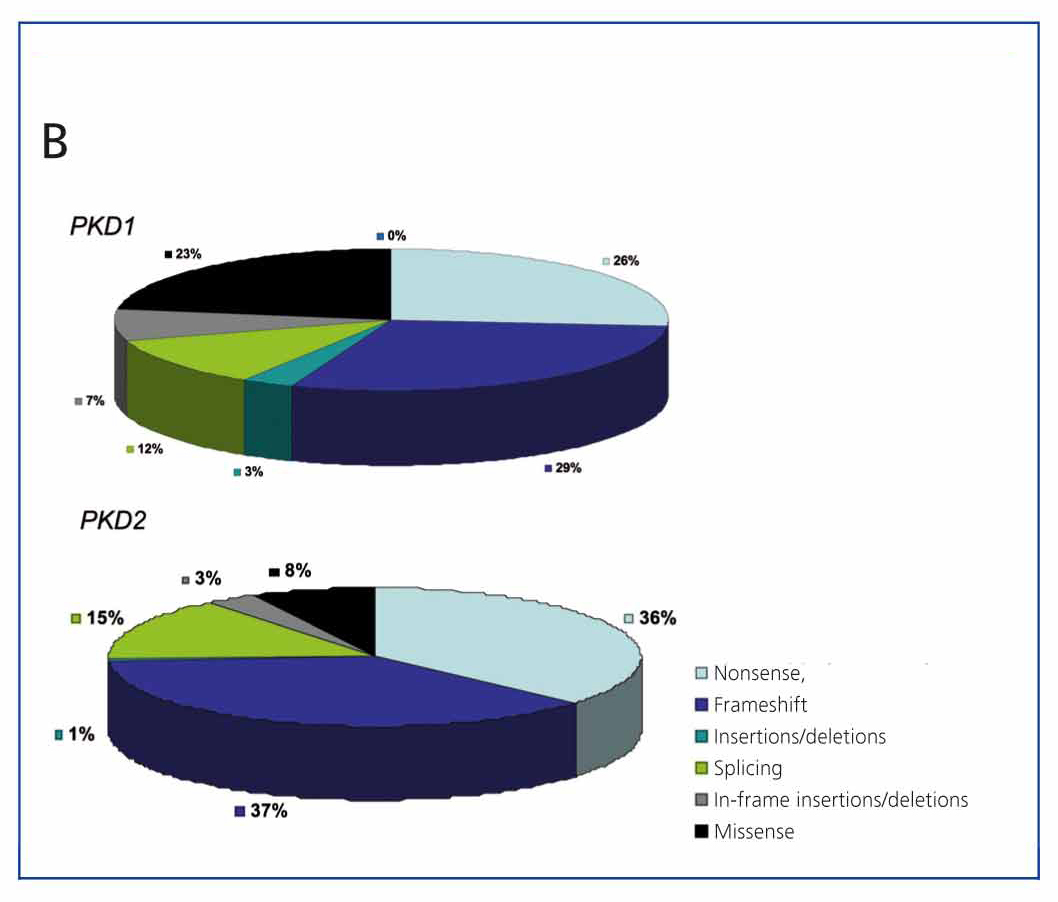
A very useful tool for this is the database developed and maintained by the Mayo Clinic (http://pkdb.mayo.edu/cgi-bin/mutations.cgi), which includes all the sequence variants described in these genes. Table 1 shows the types of mutations that cause ADPKD. As can be seen in Figure 3A, there are mutations that are clearly pathogenic, while a considerable percentage of sequence variants identified in these genes are probably neutral. Variants expected to result in a truncated protein (smaller than the wild-type protein), and those that affect canonical splicing sequences that are 100% conserved, are generally considered to be clearly pathogenic and require no further study. On the other hand, the in-frame variants that do not interrupt the translation process of the protein (amino acid change variants, deletions/insertions of base numbers which are multiple of 3, mutations in non-coding regions) require further evaluation. If we add to this the fact that each patient has an average of 10 neutral variants in the PKD1 gene, we can begin to see how difficult diagnosing the disease is in many cases. There are bioinformatic tools used to classify these sequence variants, thus determining their pathogenicity.12,25-28 To date, there are 436 different PKD1 mutations and 115 for PKD2 on the database. Figure 2B shows the different types of sequence variants identified for both PKD1 and PKD2. Sometimes, there are large deletions that remove up to 10 genes near the 5’ region without phenotypic consequences, apart from ADPKD. Moreover, deletions may occur in the 3’ region which also includes the TSC2 gene (which causes one of two types of tuberous sclerosis), giving rise to a contiguous gene syndrome which clinically results in early onset polycystic kidney disease and tuberous sclerosis. As can be seen, the percentage of sequence changes likely to be highly pathogenic is much higher in PKD2 than in PKD1 (Figure 2A). Likewise, the percentage of missense mutations (change of direction) is much higher in PKD1, which makes diagnosis difficult as their pathogenicity must be proved (Figure 2B).
In total, sequence variants with a high probability of pathogenic mutations can be identified in 91% of families. Of these, 65% are protein-truncating mutations and can therefore be directly used for diagnosis, but 26% are in-frame variants that require careful analysis before they can be used in testing.29 The segregation of these in-frame variants in a given family is a great help in definitively establishing their pathogenicity. It is also very important to consider whether they alter highly conserved amino acids in homologous proteins. Also, the inclusion of these mutations in databases will give a more accurate determination of pathogenicity when the same mutations are found again in other families.
Instead of or in addition to bioinformatic tools, functional studies can be performed for other genes to determine the functional consequences of sequence variation, and therefore, their pathogenicity. Although this is theoretically possible for PKD2 and for some PKD1 mutations, these are extremely time-consuming techniques, which are not sufficiently sensitive or specific and not applicable for routine diagnosis. Therefore, although a functional test would be helpful, bioinformatic tools are likely to be much more important in the genetic diagnosis of ADPKD than functional studies.
Additional difficulties with genetic diagnosis by detection of mutations in autosomal dominant polycystic kidney disease.
Individuals with autosomal dominant polycystic kidney disease with no mutation detected
No pathogenic mutation in either PKD1 or PKD2 is found in about 9% of patients with ADPKD.29 These patients usually have a milder form of the disease, and usually do not have a family history of the disease. Thus, the non-detection of mutations is not only a lack of sensitivity of the technique but could in fact be indicative of another disease. Firstly, there may be changes in the introns that affect splicing (which could be detected by intron sequencing or mutation studies using RNA). There may be mutations in the regulatory regions, such as the promoter (so far no mutations in these regions have been described); or there might be changes that may alter the protein assessed as non-pathogenic in silico (using bioinformatic tools), that may, however, produce a subtle alteration of the protein causing the disease. Secondly, families have been described with no linkages to PKD1 or PKD2 although, after fully exploring these families, there are doubts about the linkage analysis being correct.30 Finally, there are cystic diseases with a similar phenotype/clinical course which can behave like phenocopies, due to the genes HNF1B, PRKCSH, SEC63 or PKHD1.
Mosaicism
Mosaicism (the coexistence of normal and mutant cell populations in an individual) is common in genetic diseases with a high rate of de novo mutations. Only 10% of cases are de novo in ADPKD, so this phenomenon is not expected to be very common. Mosaicism can be germinal (when it affects only the germline), somatic (if the genetically distinct cell populations are only somatic) or gonosomic (when it involves both somatic and germ cells).
Two ADPKD families with mosaicism have been described.31,32 To detect it, the first generation of the family with one or more relatives affected by the disease (de novo case) has to be available. Mosaicism may explain the large phenotypic variability within a family, and it must be taken into account when offering genetic counselling. Thus, a patient with apparently healthy parents, and therefore considered a de novo or sporadic case, may have affected siblings if one parent suffers from germinal mosaicism (having germ cells with and without the mutation). The presence of mosaicism is one of the reasons why genetic linkage analysis is not conclusive and may be negative for PKD1 and PKD2. In addition, when there is somatic mosaicism, there may be different levels of a mutant allele in different tissues. Therefore, a determination in peripheral blood may not be representative of what is happening in the kidneys.31
Genotype-phenotype correlation and hypomorphic alleles
Current data shows poor genotype-phenotype correlation for both PKD1 and PKD2. Although some reports show some correlation for a mutation type or location, the results are insufficient to establish a prognosis based on the detected mutation.33,34
It was believed that missense mutations in the PKD genes were inactivating. However, recently28,35 it was seen that some of these changes result in hypomorphic alleles or incomplete penetrance that behaves as if the ADPKD were a recessive disease. This phenomenon has also been observed in animal models of ADPKD.36
Recently, members of families with ADPKD have been detected who are affected very differently by the disease, due to the presence of hypomorphic alleles.28,35 Thus, individuals with the two hypomorphic PKD1 alleles in trans have a severe phenotype, whereas individuals in the family with only one of these alleles have a much milder form of the disease, or may not even have detectable kidney cysts. As a result, the disease may be erroneously labelled in these families as recessive polycystic kidney disease or as sporadic cases of ADPKD. In both cases, the error may have serious consequences for genetic counselling and will result in linkage analysis errors. To determine the pathogenicity of these hypomorphic alleles resulting in an amino acid change in polycystin 1, the available bioinformatic tools must be consulted.
It is estimated that between 43% and 50% of the clinical variability in ADPKD, regarding the age of patients with end-stage kidney disease (ESKD), stems from modifying genetic effects.37-39 It is not currently known to what extent the hypomorphic alleles are involved in the phenotypic variability of ADPKD.
The current status of genetic diagnosis of autosomal dominant polycystic kidney disease
In general, genetic testing for ADPKD is not recommended when the clinical and imaging diagnosis is clear, because it is very expensive and in many cases, does not provide relevant information. Table 2 summarises the current indications for genetic diagnosis of ADPKD.
Genetically testing patients to determine whether the gene causing the disease is PKD1 or PKD2 is questionable, as there is a significant clinical variability within each gene,40 although it is clear that being a mutant PKD2 gene carrier leads to a better outcome than for PKD1.41
The genetic diagnosis of ADPKD is not extensively indicated due to the cost and current technical complexity of determining whether the changes detected in the DNA sequence are actually pathogenic mutations. As sequencing techniques become cheaper and the pathogenicity of the detected changes can be better determined, the technique may be used much more widely.
Moreover, there is currently no effective treatment for ADPKD, although there are several ongoing clinical trials. It is hoped that in a few years there will be treatment for the disease, and then, it will be necessary to be sure about the diagnosis of a patient. Therapies may be initiated in younger patients where imaging diagnosis is inconclusive.
CONCLUSIONS
Direct genetic diagnosis of ADPKD is now possible in Spain, although the analysis is not easy or cheap due to the characteristics of the PKD1 gene. Each case must be considered individually to determine whether genetic testing should be carried out and what type of study is appropriate. This type of diagnosis is of particular interest to living donors and in neonatal and sporadic cases. Genetic testing offers the chance of performing prenatal or pre-implantation testing in families with severe cases of the disease. Also, this will enable the disease to be treated, when specific treatment becomes available, in unconfirmed cases that would not be candidates for treatment if it was not for genetic confirmation.
KEY CONCEPTS
1. Molecular diagnosis of ADPKD is increasingly feasible and provides increasing information.
2. The most important uses are currently determining whether a living donor candidate has the disease or not, and for couples who want a pre-implantation genetic diagnosis.
3. Genetic studies confirm the suspected diagnosis of the disease in sporadic patients or those with atypical symptoms.
4. It is of interest in determining if a family is PKD1 or PKD2, as this is more indicative of the clinical course of the disease in the family than the genotype itself.
5. Hypomorphic alleles significantly alter the PKD1 phenotype and have important significance in prognosis and genetic counselling.
6. When ADPKD treatment is available, a precise diagnosis of the disease must be available.

Figure 1. Structure of the PKD1 (A) and PKD2 (B) genes and their transcripts

Figure 2. Genetic linkage analysis for PKD1 and PKD2.

Figure 3a. Classification of sequence variants found in the PKD1 and PKD2 genes according to the mutation database in PKD1 and PKD2 (http://pkdb.mayo.edu/cgi-bin/mutations.cgi)

Figure 3b. Classification of sequence variants found in the PKD1 and PKD2 genes according to the mutation database in PKD1 and PKD2 (http://pkdb.mayo.edu/cgi-bin/mutations.cgi)
10727_108_12807_en_w4777107431310727tabla_1_en.doc
Table 1. Mutations in PKD1 and PKD2 that cause autosomal dominant polycystic kidney disease
10727_108_12808_en_w4777107431210727tabla_2_en.doc
Table 2. Genetic diagnosis indications for autosomal dominant polycystic kidney disease



