
INTRODUCTION
Cryopyrin associated periodic syndrome (CAPS) or cryopyrinopathy is essentially composed of three different entities that often overlap: familial cold auto-inflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal onset multi-system inflammatory disease (NOMID). Each of these are produced by mutations occurring in the same gene: NRLP3 (CIAS1), which is situated on chromosome 1q44 and codes for a protein called cryopyrin (NALP3).1 These disorders are autosomal dominant with varying levels of penetrance.
Cryopyrin forms part of the multi-protein complex known as an inflammasome, which induces the formation of interleukin 1β (IL-1β) and interleukin 18 (IL-18) through various pathways. Under normal conditions, the inflammasome is activated due to a variety of stimuli, such as molecular patterns associated with certain pathogens like viral or bacterial RNA or by molecular patterns associated with tissue damage, such as uric acid crystals or cutaneous irritants.2
It is believed that in CAPS, point mutations to the NALP gene promote aberrant inflammasome formation and inappropriately elevated production of IL-1β. It is also believed that the different mutations have different impacts on inflammasome activity, which, together with the genetic qualities of each individual, would produce the different clinical entities that can be observed.3
CASE REPORT
Our patient was a 41-year-old female, ex-smoker, with a history of gastric ulcers and gastrointestinal bleeding secondary to the intake of non-steroidal anti-inflammatory drugs. She also had diffuse multi-nodular goitre that had been diagnosed two years prior, a biopsy compatible with AA amyloidosis, stage 3 chronic kidney disease (CKD) of four years evolution, and recurrent conjunctivitis with bilateral glaucoma secondary to corticosteroid abuse. Her history of surgical interventions included tonsillectomy, bilateral trabeculoplasty for glaucoma, and right oophorectomy due to ovarian cyst torsion. From 6 months, the patient had suffered episodes of cold-induced hives of 24-48 hours evolution (attacks peaked at 2-3 hours) producing wheals that left residual erythematous marks with painful burning sensation. From 11 years, the patient also had bilateral arthritis (knees, ankles, elbows, and interphalangeal joints), conjunctivitis, chills, and a sensation of fatigue and dysthermia, especially during the winter and triggered by cold temperatures, air conditioning, stress, and menstruation. The most recent episodes had also been accompanied by abdominal discomfort, nausea, and anorexia. Symptoms required prolonged rest and slowly dissipated with the help of antipyretics and corticosteroids. In recent years, symptoms had become more intense and incapacitating, with increased frequency of episodes that overlapped until symptom-free periods were very scarce. In addition, the patient had started to lose hearing in recent years (audiometric findings: bilateral perceptive deafness with important decrease in acute sound perception).
Family background
Daughter with CIAS/NALP3 mutation (cold-induced familial auto-inflammatory disease). Mother, uncle, and maternal grandfather with symptoms of hives, arthralgia-arthritis, conjunctivitis, and cold-associated influenza-like syndrome, and grandfather with renal failure.
Complementary studies revealed: creatinine: 2.2mg/dl; urea: 54mg/dl; creatinine clearance: 31.4ml/min/1.73m2; albumin: 3.8mg/dl; IgM: 384mg/dl; C4: 14.1mg/dl; haemoglobin; serum amyloid A: 21.8mg/l. Total protein, total cholesterol, liver function tests, creatine phosphokinase, IgA, IgG, C3, and thyroid hormone levels were all normal; antinuclear antibodies were negative. Urine sample: 5-7 leukocytes/field; proteinuria: 180mg/24 hours. Echocardiogram: no abnormalities. Abdominal ultrasound: normal size kidneys with diffuse increase in echogenicity.
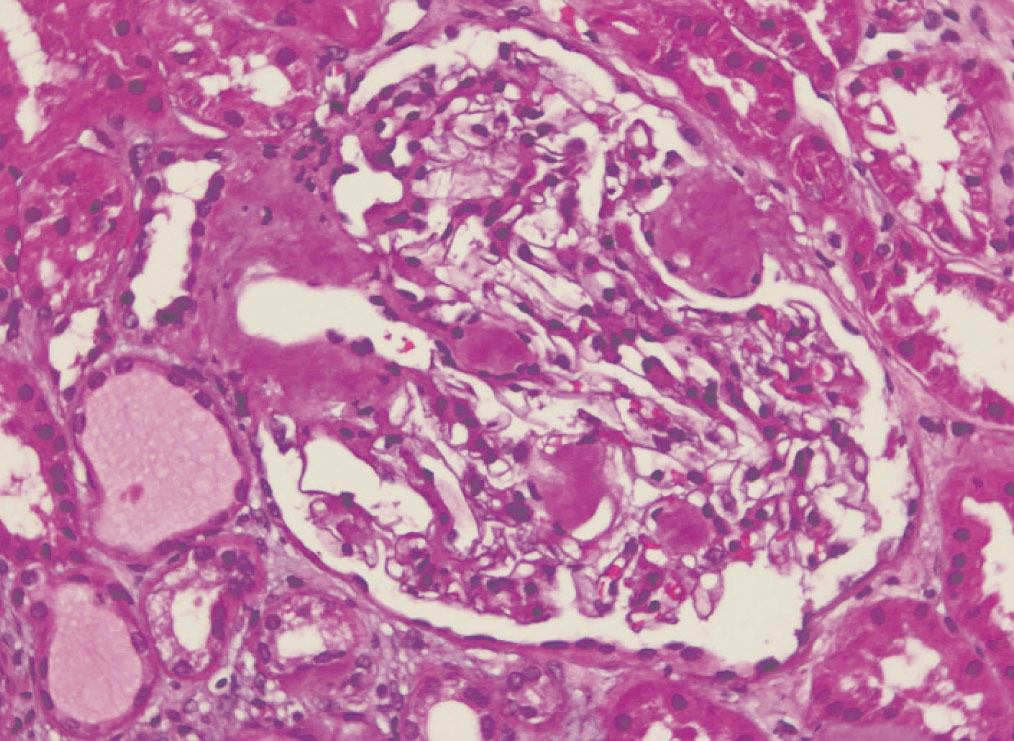
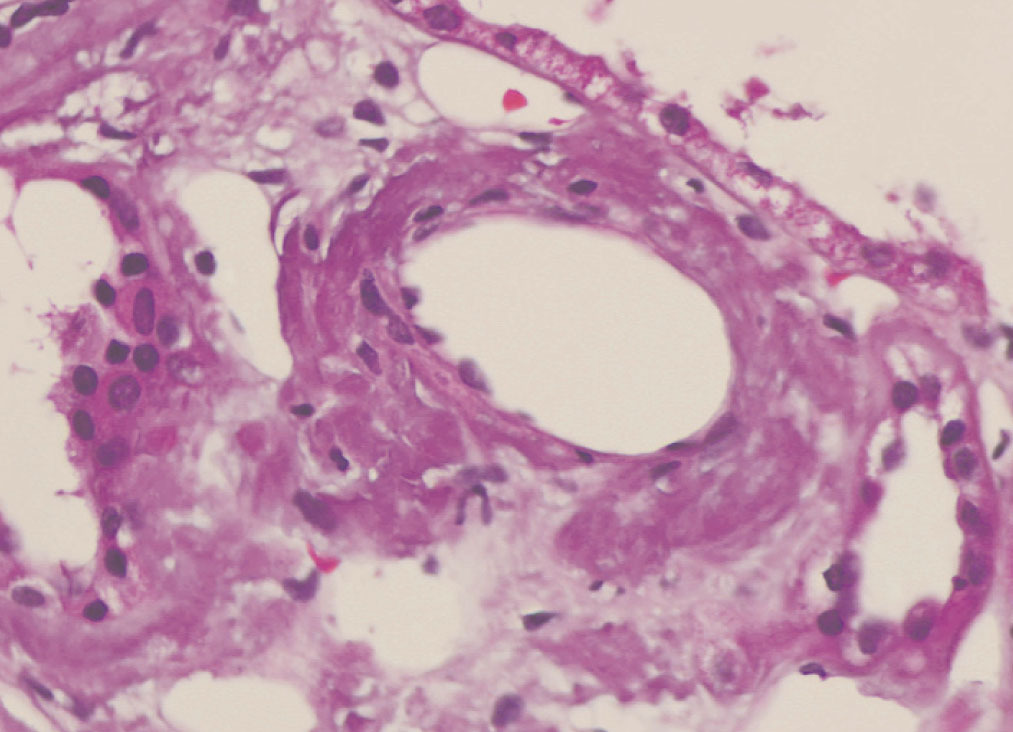
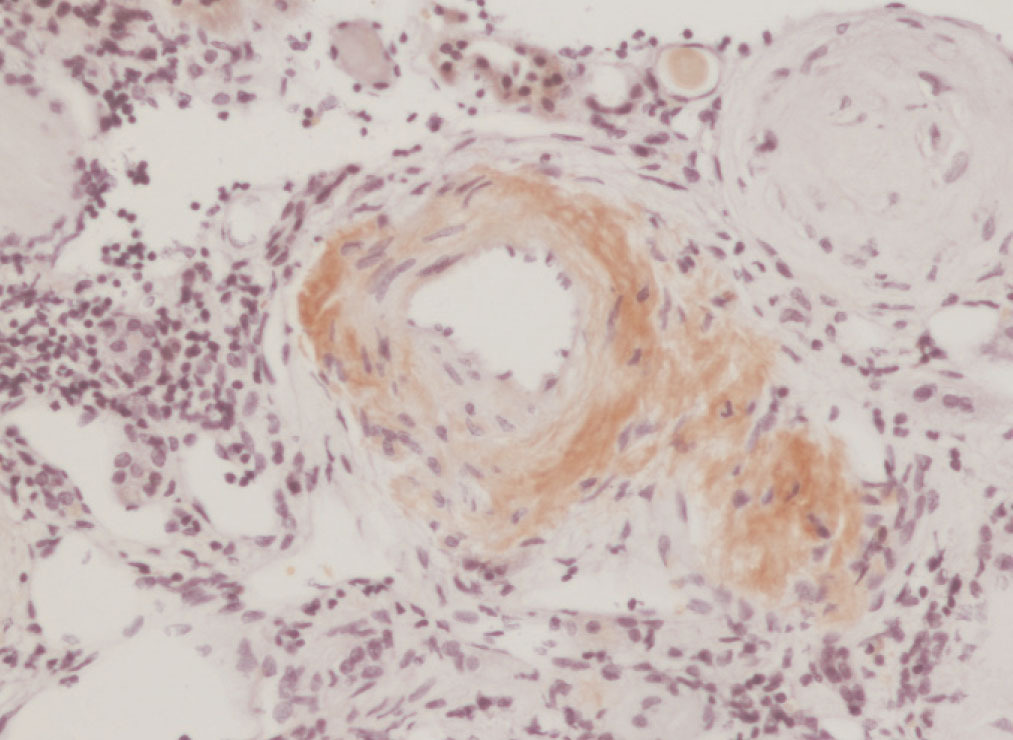
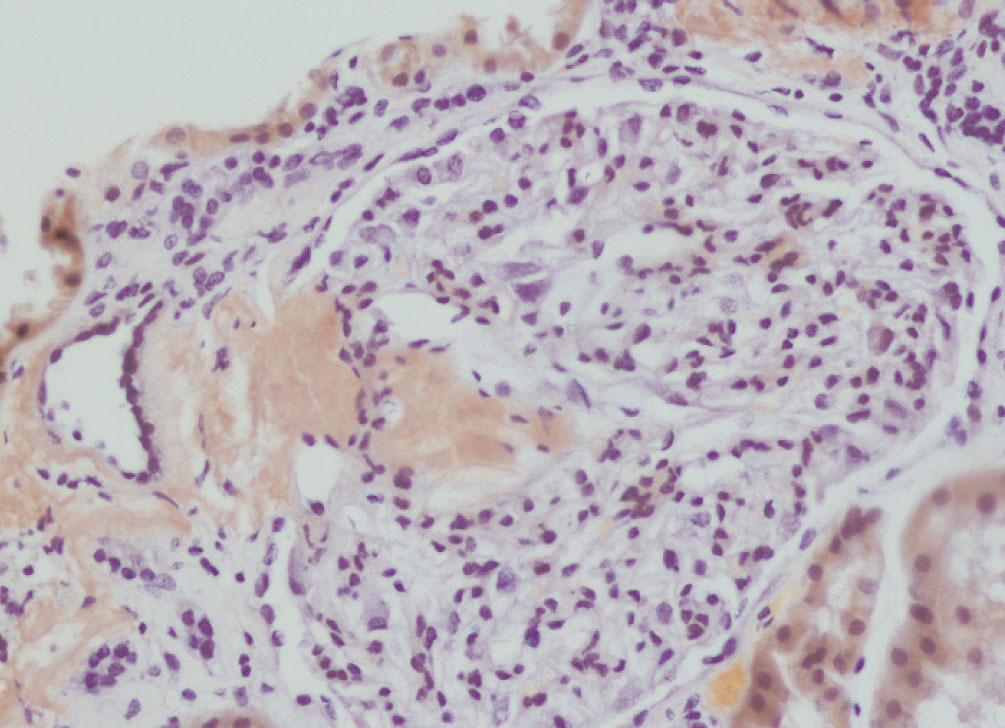
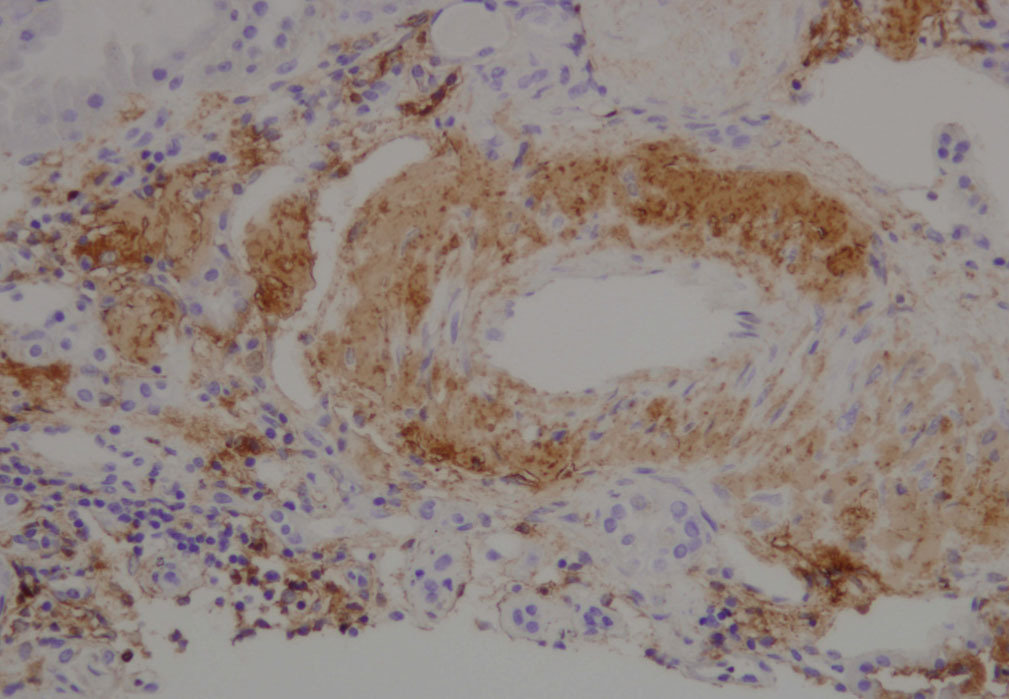
We decided to perform a renal biopsy, which showed that 17 of 26 glomeruli were sclerosed. In some of the non-sclerosed glomeruli, we observed very focal nodular deposits of hyaline eosinophilic, acellular material with weak periodic acid signatures (PAS+), more frequently found in the vascular pole (Figure 1). This material was also observed in greater quantities in arterial and arteriolar walls and interstitium (Figure 2). This material was stained with Congo red stain (Figure 3 and Figure 4), which disappeared after treatment with permanganate, and had birefringence of an apple-green colour with polarised light. Using immunohistochemical techniques, the acellular material showed staining patterns of amyloid A (Figure 5). We also observed interstitial fibrosis and moderate tubular atrophy (20%). With these histological findings, we diagnosed the patient with secondary renal amyloidosis (AA) with glomerular, vascular, and interstitial involvement.
The patient underwent a total thyroidectomy at another hospital, and a histological evaluation demonstrated the presence of abundant amyloid deposits amidst the thyroid follicles.
DISCUSSION
This clinical/pathological case was compatible with a diagnosis of reactive amyloidosis (thyroid and renal) and secondary renal failure as a complication of cryopyrin associated periodic syndrome (Muckle-Wells syndrome) (mutation NLRP3). During acute episodes, this entity has been described to progress with severe inflammation, leukocytosis, and elevated concentrations of acute-phase proteins, such as C-reactive protein and serum amyloid A (SAA).4
Reactive amyloidosis is a severe complication of CAPS (occurring in approximately 25% of cases of Muckle-Wells)5 and presents a similar pathophysiology to other types of secondary amyloidosis. This complication is caused by extracellular deposits of fibrils in the body’s tissues, which are polymers of the precursor protein SAA. Specific modulators for the risk of developing amyloidosis have been described, such as concentrations of SAA, polymorphisms of the SAA gene, and ethnic origins. Amyloidosis with renal involvement tends to progress with nephrotic range proteinuria; however, in our case, this metric was normal, which may have translated into a reduced deposition of amyloid material in the glomeruli, thus producing a less severe alteration to the filtration barrier.
Amyloidosis is defined as the extracellular accumulation of a protein with positive Congo red stain tests, apple-green birefringence test results with polarised light, and ultrastructural examination results revealing unbranched fibrils of 10nm in a random configuration. The amyloid material may be composed of a variety of different polypeptides, which can include immunoglobulin light chains (AL amyloid), heavy chains (AH amyloid), protein A amyloid (AA amyloid), beta-2-microglobulin, transthyretin, procalcitonin, beta-amyloid protein, cystatin C, etc. Secondary (AA) amyloidosis is associated with several different medical entities, such as chronic autoimmune diseases and chronic infectious diseases, but can also appear as a complication from familial Mediterranean fever and CAPS. Regardless of cause, the amyloid material always presents the same histological and ultrastructural characteristics.6 In our case, the renal biopsy revealed typical characteristics of AA type secondary amyloidosis with primarily vascular deposits of amyloid material, but also in the glomerulus and interstitium, with apple-green birefringence in polarised light that was lost after treatment with permanganate. Positive immunohistochemical tests for amyloid A confirmed diagnosis.
The patient has been on treatment for approximately 2 months with canakinumab, a monoclonal anti-IL-1β antibody, which has produced a complete disappearance of clinical symptoms, and is under surveillance to monitor the evolution of renal function and hearing.
QUESTIONS
1)Dr. Miguel Ángel Frutos (Málaga): It is surprising that this patient would have severe amyloid AA deposits in the renal biopsy yet show no evolution in proteinuria. What explanation do you give for this finding?
Response (Juan Mosquera): The deposition of amyloid material in this case was primarily vascular and interstitial, with little deposition in the glomeruli (only found in two of the glomeruli biopsied), which could explain the lack of proteinuria.
2) Dr. Francisco Rivera (Ciudad Real): I agree with the affirmation that renal amyloidosis can progress with minimal proteinuria when the deposits are exclusively vascular or interstitial. Several years ago, we published a case of type AA vascular amyloidosis associated with Still’s disease that progressed with minimal proteinuria and renal deterioration.7
Response (Luis Bolaños): In both AL and AA forms of renal amyloidosis, deposits are primarily in the form of glomerular fibrils, which in turn lead to significant proteinuria. However, much less common is the form observed in our case, in which the amyloid fibril deposits were primarily situated in the blood vessels, causing them to narrow.8 In these cases, the pattern of renal involvement is different, with progressive CKD and little to no proteinuria. Several biochemical analyses have suggested that the location of the deposits might be determined at least in part by the size of the amyloid A fragments, with smaller fragments predominately lodging in the glomerulus and larger fragments in the blood vessels.9
3) Dr. Rafael Poveda (Barcelona): You have presented on a rather uncommon disease with secondary amyloidosis. Did you at any point try treatment with colchicin?
Response (Luis Bolaños): Colchicin is a widely accepted form of treatment for preventing secondary amyloidosis in familial Mediterranean fever. In these cases, colchicin reduces the frequency of episodes of abdominal pain and the incidence of kidney disease, and stabilises renal function in patients with mild proteinuria.10 In other clinical contexts, such as inflammatory bowel disease and Behcet’s disease, its use is anecdotal. We do not know if its mechanism of action is due to a direct reduction in the production of amyloid fibrils or rather due to decreased activity of the primary disease.
In our case report, treatment with canakinumab produced a complete response and control of the inflammatory process, and as such, of the stimulus for producing new amyloid material, thus obviating the need for treatment with colchicin.
4) Dr. Manuel Solé (Barcelona): My comment is related to the presence of hidden and unexpressed inflammatory states that are difficult to diagnose and thus are usually diagnosed late. Their effects, as in this case, are recognised only in advanced stages of CKD. What laboratory tests should always be applied in these cases?
Response (Luis Bolaños): The three known forms of CAPS (FCAS, MWS, and CINCA [chronic infantile neurological cutaneous and articular syndrome]/NOMID) are very uncommon and have their origins in mutations to the NLRP3 gene. In the two first of these conditions, inheritance of the genetic mutation is autosomal dominant and more rarely sporadic, whereas in the last the majority of cases are sporadic and rarely autosomal dominant.
In all of these, the necessary clinical symptoms (primarily episodic inflammation) and the presence of family members with similar symptoms must alert us to the possibility of one of these types of diseases. As a second step, a genetic analysis should be carried out in order to determine what type of mutation has occurred to the NLRP3 gene, whether in the patient in question or in affected family members, which would indicate the most probable phenotype. Finally, if MWS is suspected, a bilateral audiogram should be administered and several laboratory parameters should be evaluated: serum levels of protein A and C-reactive protein, parameters of renal function and proteinuria, a renal biopsy in the case of renal failure and/or significant proteinuria, and a thorough examination of the presence and severity of auditory and renal involvement.
5) Dr. Vicente Barrio (Madrid): Did you perform any background genetic analyses in this case?
Response (Luis Bolaños): We did not administer any genetic tests on our patient; however, the patient’s daughter, of 5 years of age, had undergone these tests, which revealed a mutation in the NLRP3 gene.
The different mutations described for the NLRP3 gene associated with CAPS can be associated with single phenotypes: FCAS, MWS, or CINCA/NOMID, or with multiple phenotypes: FCAS-MWS or CINCA/NOMID-MWS. In children with a defined mutation associated with FCAS-MWS phenotypes, the clinical symptoms at that moment may simply be repeated inflammatory syndromes, with the possibility of developing other complications in more advanced stages of disease. In the case of our patient’s daughter, the only symptoms were those of repeated inflammatory syndromes. Despite the lack of long-term experience with anti-interleukin 1, early treatment potentially could allow for preventing severe and irreversible hearing loss and kidney damage that might occur later in life in MWS were allowed to develop.
6) Dr. Rafael Poveda (Barcelona): In addition to the amyloid renal involvement, I was interested by the presence of deafness. Do these two manifestations share a common source of tissue damage?
Response (Luis Bolaños): Sensorineural hearing loss in MWS is bilateral, progressive, and can occur in adolescence or adulthood, with the moment of appearance being similar in members of the same family.11 Both in MWS and in CINCA/NOMID, cochlear inflammation arises with no concomitant appearance of amyloid deposits in the Corti apparatus or cochlear nerve. Autopsy studies on patients with these syndromes have revealed atrophy of the cochlear nerve and an absence of the Corti apparatus, which suggests that the inflammatory processes provoke irreversible damage on these structures, lending even greater importance to early identification and treatment.12,13
7) Dr. Rafael Poveda (Barcelona): Have any other systemic manifestations of MWS been described?
Response (Luis Bolaños): This entity was first described in a British family in 1962 by Muckle and Wells. In the scientific literature, in addition to the classic triad of recurrent auto-inflammatory episodes (fever, cutaneous rash, arthralgia, abdominal pain, and conjunctivitis), sensorineural deafness, and secondary amyloidosis, other alterations have also been described: mono- and oligo-articular arthritis (primarily in the knees, ankles, shoulders, and small joints of the extremities), chronic headache, and irritability secondary to increased intracranial pressure due to chronic aseptic meningitis of mild intensity, demyelinating lesions,14 cutaneous lesions (aphthosis, ichthyosis, hypertrichosis, and hyperpigmentation), delayed growth, and mild mental retardation.15
8) Dr. Miguel Perdiguero (Alicante): Would serial measurements of AA amyloid substance during treatment with canakinumab provide data in terms of a response marker?
Response (Luis Bolaños): AA amyloid material is the precursor protein to secondary or reactive amyloidosis. This is an acute phase protein, primarily produced in the liver due to stimulation by proinflammatory cytokines such as interleukin 1. During active inflammation, the concentrations of this protein in serum samples can reach 1000mg/l, which is 1000 times greater than normal.16,17 Since patients with CAPS produce excessive levels of inflammatory cytokines (IL-1β, IL-18), the presence of AA amyloid substance within normal limits would indicate proper control of the inflammatory process, and as such, provides an appropriate marker for response to treatment with the various types of anti-interleukin 1 agents.
9) Dr. Miguel Perdiguero (Alicante): In any case, my comment is also directed towards the possible loss in effectiveness of monoclonal treatments due to the presence of antibodies against biological agents, and with this, a loss of efficacy and the need for switching to other treatment alternatives.
Response (Luis Bolaños): Among the various possible adverse reactions to biological agents, the production of neutralising antibodies falls within the beta group, which comprises the development of immunogenicity against the therapeutic protein and the possible partial or total reduction in the efficacy of the drug. The production of antibodies against a specific biological agent depends on the level of humanisation of the monoclonal antibody and the form of administration (subcutaneous, intravenous, or intramuscular), and the frequency of administration. In the case of human or humanised antibodies, which in principle are less immunogenic, it is also possible to develop anti-idiotypic IgG or IgE antibodies.18
In the particular case of canakinumab, this is a humanised monoclonal antibody against IL-1β that is applied every 8 weeks subcutaneously or intravenously, which has been approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2009 for use in the treatment of FCAS and MWS. There has not been a great deal of experience gained in treating patients with this drug, and until now no studies have described the appearance of neutralising antibodies. In the hypothetical case of loss of efficacy due to neutralising antibodies, the currently available alternatives are: rilonacept, approved for use in FCAS and MWS in patients older than 12 years (FDA, 2008 and EMA, 2009) and anakinra, which has been used to treat CAPS outside of the indications on the drug technical data sheet with good results.
Acknowledgements
We would like to thank Novartis for their continued collaboration with the organisation of the GloSen conference and Nephropathology Club.
Conflicts of interest
The authors declare that they have no conflicts of interest related to the contents of this article.

Figure 1. Glomerular amyloid deposits with haematoxylin-eosin stain

Figure 2. Arterial amyloid deposits with haematoxylin-eosin

Figure 3. Congo red stain in the glomerular vascular pole

Figure 4. Arterial amyloid deposits

Figure 5. Amyloid A deposits using immunohistochemistry



