

INTRODUCCIÓN
La enfermedad de Fabry es un trastorno hereditario del catabolismo de los glicoesfingolípidos producido por el déficit del enzima lisosomal α-galactosidasa A (α-GAL A), que origina el depósito intracelular, especialmente globotriaosilceramida (Gb-3), en el endotelio vascular y otros tejidos. Se transmite ligada al cromosoma X, y hasta la actualidad han sido descritas más de 400 mutaciones (Human Gene Mutation Database, Institute of Medical Genetics, Cardiff http://archive.uwcm.ac.uk/uwcm/mg/hgmg0.html). Tradicionalmente se ha considerado de transmisión recesiva, que las mujeres heterocigotas son portadoras, y que sólo un 1% desarrollaría la enfermedad debido a la inactivación al azar de uno de los cromosomas X (conocido como efecto Lyon), sin embargo, hay una evidencia creciente que un gran porcentaje de mujeres heterocigotas tiene déficits enzimáticos parciales y manisfestaciones clínicas con expresividad variable1-5.
Es una enfermedad progresiva que causa manifestaciones derivadas de la disfunción del órgano afectado por los depósitos, principalmente riñón, corazón, sistema nervioso, tracto gastrointestinal, y piel, aunque puede participar cualquier órgano y sistema de la economía4,6,7. Estudios clínicos8,9 y experimentales10 han puesto de manifiesto que la enfermedad de Fabry condiciona un estado inflamatorio vascular y protrombótico. De hecho, los eventos cardiovasculares, pricipalmente cardiopatía isquémica11 y accidentes cerebrovasculares12 son una causa importante de morbi-mortalidad en estos pacientes.
En los últimos años se ha constatado la diversidad de presentación clínica, con formas parciales de manifestación tardía diagnosticadas de manera casual o bien mediante estudios dirigidos, que han puesto de manifiesto que aunque es una enfermedad considerada «rara» por su baja frecuencia, la prevalencia es superior a la que se suponía, y por tanto, existe la sospecha que una cantidad indeterminada de familias afectas no son diagnosticadas. Por otro lado, la disponibilidad de tratamiento de sustitución enzimático (TSE) ha abierto nuevas y esperanzadoras expectativas, eso sí, acompañadas de un gran debate y un cierto confusionismo sobre el tipo y pautas de administración de las dos enzimas comercialmente disponibles.
VARIANTES EN LA EXPRESIÓN CLÍNICA
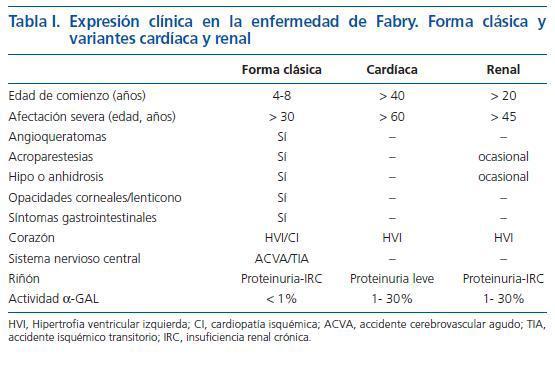
La enfermedad de Fabry se manifiesta con una gran variabilidad en el fenotipo, no sólo dentro de una misma mutación, sino incluso dentro de una misma familia13,14. Además de factores ambientales y quizá la participación de otros genes, la intensidad o grado de afectación se ha puesto en relación con la actividad residual de la enzima α-GAL A. La forma clásica de la enfermedad se suele caracterizar por el déficit absoluto de la actividad enzimática (inferior al 1%), con participación multisistémica, de comienzo en la infancia, para llegar a la afectación severa en la tercera o cuarta década de la vida4,7,13,14 (tabla I). Defectos enzimáticos parciales (1-30%) dan lugar a formas incompletas, de comienzo tardío (a partir de los 20-30 años), con afectación predominantemente cardíaca y/o renal, y escasez o ausencia de las manifestaciones clásicas de la enfermedad. En este sentido, han sido descritas la «variante cardíaca» en la que predominaría la hipertrofia ventricular izquierda (HVI)15 y la «variante renal» manifestada por proteinuria e insuficiencia renal progresiva, a menudo acompañada también de HVI (16-18) (tabla I).
En el riñón se produce depósito de Gb3 en los podocitos, mesangio, endotelio del capilar glomerular, epitelio tubular, células endoteliales y de la capa muscular de arterias y arteriolas, y en las células intersticiales19-21. Los depósitos son progresivos y conducen a la glomeruloesclerosis y fibrosis intersticial19,20. Los datos iniciales de afectación renal son la isostenuria, microalbuminuria y ocasionalmente signos de disfunción del túbulo proximal; posteriormente aparece proteinuria que en el 20% de los casos puede ser superior a 3g en 24 h y la insuficiencia renal, con o sin hipertensión arterial22. Por otro lado, ha sido referido que hasta un 10% de los pacientes con enfermedad de Fabry tienen asociadas lesiones glomerulares por otras causas20.
No se conoce bien la velocidad de progresión de la nefropatía desde los estadios iniciales. En la forma clásica se suele llegar a la enfermedad renal crónica (ERC) severa (grado 5 de NKFDOQI) entre la tercera y quinta década de la vida22,23, mientras que en las formas incompletas puede ocurrir en edades avanzadas15,18. La progresividad de la afectación renal se ha relacionado con el grado de déficit enzimático22. Una vez instaurada la insuficiencia renal, la progresión a un estadio severo puede ser rápida, similar a la nefropatía diabética. En la serie de Branton y cols. se decribe un subrupo de 14 pacientes, en los que se conocía la evolución de la función renal y que llegaron a diálisis, que tenía una tasa media de pérdida de filtrado glomerular de -12 ml/min/1,73 m2 por año, una vez alcanzada la creatinina sérica de 1,5 mg/dl22. En otra serie de 447 pacientes (62% varones) el tiempo medio en doblar la creatinina cuando se partía de un valor de 1,5 mg/dl fue de 39 meses24.
EPIDEMIOLOGÍA Y DIAGNÓSTICO
Se estima que la incidencia de la forma clásica es de 1 de cada 40.000-60.000 varones nacidos vivos (aproximadamente 0,002%)25, si bien, se desconoce la incidencia global en ambos sexos, que incluiría formas incompletas de comienzo tardío, tanto en varones como en mujeres. Recientemente ha sido publicado un estudio Italiano sobre 37.104 varones neonatos consecutivos, en el que 12 (0,03%) fueron diagnosticados de enfermedad de Fabry, que no era previamente conocida en las familias, y que podían corresponder a formas tardías26, lo que pone de manifiesto que las variantes incompletas son más frecuentes que la forma clásica de la enfermedad. De hecho, estudios prospectivos han reflejado que la enfermedad de Fabry está presente en el 3-4% de los pacientes con HVI15,27, y en el 5% de una serie de pacientes con accidentes cerebrovasculares agudos de etiología desconocida28.
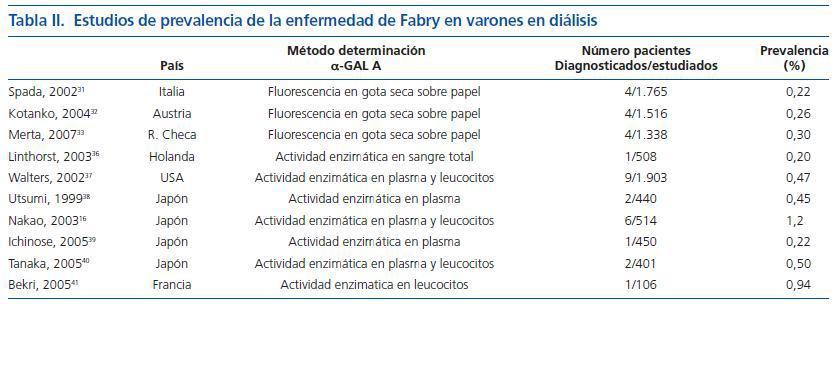
La prevalencia de la enfermedad de Fabry en los pacientes con afectación renal, se basa principalmente en el estudio de los pacientes sometidos a tratamiento renal sustitutivo. Mientras que los registros europeo y americano muestran una prevalencia del 0,018% y 0,016% respectivamente (un 12% en ambos registros eran mujeres), estudios dirigidos en pacientes sometidos a diálisis han demostrado una prevalencia muy superior. Cuando para el despistaje inicial se determinó la actividad de α-GAL Amediante fluorescencia en sangre seca sobre un filtro de papel, la prevalencia de varones en diálisis diagnosticados de novo de enfermedad de Fabry fue del 0,22-0,30%31-33 (tabla II). El principal problema del método de la gota seca es la existencia de falsos negativos34 especialmente en mujeres35. Con la cuantificación de la actividad enzimática en sangre total, plasma y/o leucocitos, estudios llevados a cabo en USA, Europa y Japón, sitúan la prevalencia de la enfermedad en varones en diálisis entre el 0,20 y el 1,2%16,36-41 (tabla II). Las tres series publicadas que incluyen a las mujeres muestran una prevalencia del 0%32, 0,05% y 0,33%40. Los datos anteriores ponen de manifiesto que la prevalencia de la enfermedad de Fabry en varones en diálisis es superior entre 15 y 80 veces a la esperada según los registros. Estos pacientes no diagnosticados antes del inicio de la diálisis, a menudo presentaban formas incompletas, con pocas o ninguna manifestación clínica extrarrenal de la enfermedad salvo afectación cardíaca, principalmente HVI16,31.
No se conoce la prevalencia de la enfermedad de Fabry en pacientes con ERC no sometidos a tratamiento renal sustitutivo. Sin duda el diagnóstico precoz tendría una gran relevancia por varias razones. Por un lado, por la posibilidad de la aplicación de un TSE, que puede evitar o retrasar la progresión de la enfermedad, y por otro, por permitir la realización del estudio familiar que conduzca al diagnóstico precoz y al consejo genético. En este sentido, se está realizando en España un estudio multicéntrico, dirigido a varones con ERC grado 1-5 (no sometidos a diálisis) de etiología desconocida, en el que como método de despistaje inicial se mide la actividad plasmática de la α-GAL A42. Resultados preliminares sobre 229 pacientes arrojan una prevalencia del 0,9%. Se trata de 2 varones de 25 y 74 años de edad, ambos con formas incompletas, con actividad de la α-GALA > de 1% y ausencia de las manifestaciones clínicas clásicas.
Por tanto, es necesario diagnosticar a los pacientes con enfermedad de Fabry lo más precozmente posible. En la forma clásica el complejo sintomático multisistémico puede alertar en la infancia, aunque con frecuencia el diagnóstico se hace en la segunda o tercera década de la vida por biopsia renal y/o la determinación en sangre de la actividad α-GAL A, con la confirmación mediante estudio genético. Las formas incompletas que llegan a Nefrología son más difíciles de detectar sino existen programas establecidos, dado que, según la experiencia de los trabajos publicados, estos pacientes por sus características pueden no ser biopsiados16,31-33,36-41.
TRATAMIENTO
El alivio de los síntomas, la reducción del daño tisular y la prevención de las complicaciones tardías son los objetivos terapéuticos fundamentales del tratamiento en la enfermedad de Fabry34,43-47. Desde el punto de vista renal, las acciones irán encaminadas a prevenir la nefropatía o a evitar o enlentecer su progresión mediante una intervención lo más precoz posible, para lo que contamos con un TSE, además de las medidas generales de prevención de la ERC.
Desde el año 2001 se dispone de dos enzimas humanas recombinantes, la agalsidasa alfa producida a partir de fibroblastos humanos (Replagal®, Shire Human Genetic Therapies, Inc), y agalsidasa beta (Fabrazyme®, Genzyme Corp) producida a partir de células de ovario de hamster chino. Estudios en fase I/II48,49 condujeron a que desde su comercialización, según ficha técnica, agalsidasa alfa se prescribe a dosis de 0,2 mg/kg en infusión cada 14 días, y agalsidasa beta a dosis de 1 mg/kg también en infusión cada 14 días, con un coste por paciente/año similar (en torno a 210.000 ¿ para un individuo de 70 kg). Varios estudios clínicos han evaluado la eficacia y seguridad de ambas formulaciones, y se han generado dos tendencias con partidarios a favor de una u otra estrategia terapeútica. Ambos productos son capaces de reducir los depósitos titulares de Gb-350-51, poseer efectos favorables de la función renal50,51 y reducir la HVI52,53, si bien, los distintos criterios de inclusión y diseño de los estudios no permitían una comparación directa.
A la luz de algunas informaciones se puede inferir que agalsidasa alfa y agalsidasa beta son similares, dado que tienen la misma actividad específica por miligramo de producto administrado, determinada in vitro por el aclaramiento de Gb-3 en fibroblastos de piel de pacientes con enfermedad de Fabry54, y existe una reactividad cruzada completa de anticuerpos IgG frente a estos productos55. Recientemente Vedder y cols. han publicado el único estudio clínico comparativo cuyos resultados apoyan también la similitud entre ambas formulaciones56. De manera prospectiva y randomizada se administró agalsidasa alfa (18 pacientes) o agalsidasa beta (16 pacientes) a la misma dosis (0,2 mg/kg cada 14 días) con un seguimiento de 24 meses. No se observaron diferencias entre ambos tratamientos en ninguno de los parámetros estudiados: proteinuria, filtrado glomerular (FG), HVI, dolor neuropático, concentración de Gb-3 plasmática y urinaria, y aparición de anticuerpos IgG56.
En 2001 fueron publicados 2 estudios prospectivos, controlados y randomizados en fase 3, con agalasidasa alfa50 y agalsidasa beta51, y en los dos se hicieron estudios de extensión en fase 4, que han permitido evaluar la función renal a largo plazo. Con agalsidasa beta fueron incluidos 58 pacientes, 29 tratados con dosis estándar (1 mg/kg/14 días) durante 20 semanas, con el objetivo primario de evaluar los depósitos de Gb-3 en el endotelio microvascular renal. Se evidenció una marcada reducción de los depósitos de Gb-3 en riñón, piel y corazón51. En una extensión a 11 meses, en el que todos los pacientes recibieron TSE (el grupo placebo a partir de la semana 20), se comprobó que los depósitos renales se mantenían con un valor cercano a cero57. Estos pacientes continuaron siendo evaluados en 2 estudios de extensión a 358 y 4,5 años59. Las medias de proteinuria y FG permanecieron sin cambios significativos, aunque 6 pacientes experimentaron deterioro de la función renal. Los principales factores de progresión fueron la proteinuria > 1 g/24 h y un porcentaje de esclerosis glomerular > 50% en situación basal. Destacar que sólo 10 de los 58 pacientes tenían al inicio del estudio un FG menor de 90 ml/min/1,73 m2, de los cuales 4, experimentaron empeoramiento de la función renal y 6 continuaron en situación estable.
En el estudio fase 3 con agalsidasa alfa se incluyeron 26 pacientes, 14 tratados a dosis 0,2 mg/kg/14 días durante 24 semanas50, que se continuó en un estudio en fase 4 durante 4,5 años, en el cual, recibieron TSE todos los pacientes a partir del sexto mes61. El tratamiento no modificó la proteinuria, y la media del FG descendió significativamente al final del periodo de estudio60. No obstante, este descenso se produjo fundamentalmente a expensas de la pérdida de función renal en todos los pacientes que basalmente tenían ERC grado 3, y algunos con estadio 2.
Otros estudios observacionales ponen de manifiesto que TSE puede estabilizar la función renal en pacientes con ERC grado 2, pero no evita la progresión cuando existe ERC de grado 3 o inferior61,62. En la serie más numerosa, con 201 varones y mujeres tratados con agalsidasa alfa, se observó que en un subgrupo de 12 pacientes con ERC grado 2, la media del FG había descendido desde 83,7 hasta 71,9 ml/min/1,73 m2 en el año previo al comienzo del TSE, que se mantuvo sin cambios un año después de su inicio61. En el subgrupo de 8 pacientes con ERC grado 3, el TSE no modificó la tasa de progresión de la insuficiencia renal62. Un interesante estudio publicado recientemente muestra como en 11 varones con la forma clásica de la enfermedad de Fabry, que presentaban pérdida de función renal mayor de -5 ml/min/1.73 m2 por año, durante el tratamiento con agalsidasa alfa en pauta convencional (0,2 mg/kg cada 2 semanas), experimentaron un enlentecimiento en la progresión al pasar a pauta semanal de 0,2 mg/kg (de -8,0 a -3,3 ml/min/1,73 m2 por año, p < 0,01)63. No queda claro si este efecto beneficioso fue debido al aumento en la frecuencia de administración, al aumento de la dosis, o a ambas. En cualquier caso, estos resultados clínicos entrarían en una aparente contradicción con la observación que ni la dosis, ni la frecuencia de administración influyeron en la magnitud de reducción de Gb-3 plasmática, en un estudio randomizado que evaluó en 18 pacientes 5 pautas distintas de administración de agalsidasa alfa (0,1, 0,2 ó 0,4 mg/kg/semana, o 0,2 mg/kg cada 2 semanas, o 0,4 mg/kg cada 2 semanas)64. Es posible que el nivel de Gb-3 no sea un marcador de severidad y respuesta al tratamiento en la enfermedad de Fabry, y los propios autores señalan que son necesarios nuevos estudios para determinar la pauta de tratamiento óptima para alcanzar los mejores resultados clínicos64.
En pacientes con insuficiencia renal crónica establecida han sido evaluados los efectos renales, cardíacos y cerebrovasculares del TSE, en un ensayo prospectivo, randomizado y controlado, en el que 51 pacientes fueron tratados con agalsidasa beta y 31 recibieron placebo, con un FG medio de 53 y 52,4 ml/min/1,73 m2 repectivamente, y una mediana de seguimiento de 18,5 meses65. Las medias del FG y proteinuria no fueron distintas de manera significativa entre ambos grupos al final del periodo de estudio. Sin embargo, el grupo de pacientes tratados presentó una reducción del riesgo de aparición de eventos renales (definidos como aumento de la creatinina mayor del 33%, diálisis o trasplante), cardíacos y/o cerebrovasculares respecto al control. Estos efectos beneficiosos fueron más evidentes en aquellos pacientes con menor grado de proteinuria (< 1 g/24 h) y menor afectación de la función renal (FG > 55 ml/min/1,73 m2 ), de lo que se infiere que en la enfermedad de Fabry la proteinuria y la función renal se pueden considerar marcadores tanto de riesgo de complicaciones cardiovasculares, como de la respuesta al TSE. La importancia de la precocidad del inicio del TSE, no sólo en cuanto a la evolución de la función renal, si no también en la prevención de complicaciones extrarrenales, ha sido puesta de manifiesto por Breunig y cols., en un estudio prospectivo que incluía 23 pacientes tratados con agalsidasa beta66. Se observó que en los pacientes con FG > 90 ml/min/1,73 m2 la función renal permaneció estable, y no presentaron eventos clínicos, mientras que en los que tenían ERC grado 2-4 hubo una progresión de la insuficiencia renal y el TSE no previno la aparición de complicaciones cardio ni cerebrovasculares66.
En relación a la respuesta inmune frente a las dos enzimas, tomando como referencia los estudios en fase 3 y su extensión59,60, un 90% de los pacientes tratados con agalsidasa beta desarrollaron anticuerpos IgG59, frente al 56% con agalsidasa alfa60. Estas diferencias pueden ser debidas a la distinta dosificación y/o al método empleado, y no tanto a la diferencia entre los dos preparados, dado que existe reacción cruzada completa entre ambas55, y en un estudio comparativo con dosis iguales la tasa de seroconversiones fue similar56. No obstante, hubo un descenso en la titulación de anticuerpos con las dos formulaciones, que llegaron a ser indetectables en algunos pacientes. No está clara la importancia que tiene en la eficacia la aparición y mantenimiento de anticuerpos IgG. En los dos estudios59,60 se refiere que la seroconversión no tiene influencia, bien en base a la eliminación de Gb-3 en tejido renal59, o en base a la eliminación urinaria de Gb-3 y la evolución del FG60. Sin embargo, otros autores, encuentran que los pacientes que desarrollaron anticuerpos IgG, tenían una menor eliminación de Gb-3 en la orina, comparados con los que no seroconvertían, tanto con agalsidasa alfa como con agalsidasa beta56, lo que abre interrogantes sobre esta cuestión. En cuanto a los efectos adversos, la mayoría de los pacientes en tratamiento con agalsidasa beta y un 56% con agalsidasa alfa, presentó al menos uno durante todo el periodo de seguimiento. Estos efectos fueran en su mayoría leves, relacionados con la infusión y disminuían con el tiempo59,60.
De estos trabajos se desprende que aquellos pacientes con FG normal sin proteinuria o con proteinuria menor de 1 g/24 horas, el TSE evita la progresión de la ERC y previene la aparición de complicaciones. En los enfermos con ERC grado 2 la respuesta al TSE es más difícilmente predecible, siendo la proteinuria > 1 g/24 h un factor de peor pronóstico. La glomeruloesclerosis y el daño intersticial ya establecidos, hacen que el TSE no evite la progresión de la ERC cuando el FG es menor de 60 ml/min/1,73 m2, si bien, se justifica por la posible prevención en la aparición de complicaciones, y el alivio de algunos síntomas.
De manera generalizada se observa que la proteinuria no se reduce con el TSE50,51,56,59,60,66, a pesar de la masiva disminución de los depósitos renales de Gb-3, aun con FG normal50,51,66. Esto indica la presencia de lesiones estructurales glomerulares e intersticiales ya irreversibles desde que la proteinuria está presente. Sin embargo, el tratamiento enzimático puede reducir la microalbuminuria67 lo que es un dato más a favor de la necesidad de un inicio precoz. En cualquier caso, existe la idea que en la enfermedad de Fabry con afectación renal han de ser establecidas las medidas generales de cualquier nefropatía proteinúrica, como son la dieta, el control de la hipertensión y de la hiperlipidemia, y el empleo de inhibidores del enzima de conversión de la angiotensina (IECA)/ bloqueantes del receptor de la angiotensina (ARA II)68. Los beneficios de la terapia reductora de la proteinuria con IECA y/o ARA II han sido puestos de manifiesto en un reciente trabajo, en el que se constata descenso de la misma y estabilización de la función renal en pacientes con ERC grado 2 tratados con agalsidasa beta69.
Grupos de Expertos han elaborado Guías de evaluación y tratamiento de la enfermedad de Fabry70,71. Un punto importante es la recomendación de cuando ha de ser iniciado el TSE. En las Guías para el Estudio y Tratamiento de la Enfermedad de Fabry (GETEF) se establecen criterios mayores y menores, de tal manera que para el comienzo se precisaría 1 criterio mayor o 2 menores; entre ellos, la proteinuria ( > 300 mg/24 h en adultos o > 5 mg/kg/día en niños) y el FG < 80 ml/min/1,73 m2 son criterios mayores, mientras que la microalbuminuria está dentro de los criterios menores70. Sin embargo, otras guías recomiendan ofrecer terapia enzimática a todo varón adulto (mayor de 16 años) diagnosticado de enfermedad de Fabry con independencia del estadio de ERC, en niños cuando aparezcan síntomas, y en mujeres si hay síntomas o signos de afectación orgánica71.
En los pacientes en diálisis, el TSE estaría indicado para prevenir las complicaciones extrarrenales de la enfermedad72. La administración de agalsidasa beta durante la sesión de hemodiálisis, tanto de alto como de bajo flujo, es bien tolerada sin pérdida de la actividad enzimática73. Aunque las lesiones típicas de la enfermedad de Fabry no aparecen en el riñón trasplantado, el TSE estaría también justificado para tratar y prevenir la afectación multisistémica74.
CONCLUSIONES
En resumen, la prevalencia de la enfermedad de Fabry es superior a la referida en los registros oficiales de pacientes en tratamiento renal sustitutivo, debido a la existencia de variantes incompletas en la expresión clínica de presentación tardía, con afectación predominantemente cardíaca y renal, y ausencia de otras manifestaciones típicas. Estas formas incompletas son difícilmente diagnosticables si no es a través de programas establecidos. Dada la importancia del diagnóstico precoz, estos programas de detección tienen especial trascendencia en las consultas de Nefrología. La proteinuria superior a 1 g/24 h y/o la disminución del FG son factores pronósticos, tanto en la aparición de complicaciones cardíacas y cerebrovasculares, como de la respuesta al TSE. Este, probablemente debería ser aplicado en fases muy precoces para prevenir la aparición de lesiones estructurales renales, aunque persisten todavía interrogantes sobre cual es la pauta de tratamiento óptima en cuanto a dosificación y frecuencia de administración. En los pacientes con ERC el TSE puede frenar la progresión en estadios 1 y 2, y reduce la aparición de complicaciones cardiovasculares en estadios más avanzados. Aunque los datos disponibles son escasos, estos pacientes se beneficiarían del tratamiento con IECA/ARA II, y otras medidas generales de prevención de la progresión de la ERC.

Tabla 1.

Tabla 2.



