
In children, chronic pancreatitis (CP) is usually associated with anatomical anomalies of the pancreas and biliary tract or is genetically determined. Autosomal dominant polycystic kidney disease (ADPKD) may present with extrarenal cyst formation, sometimes involving the pancreas. Large enough, these cysts may cause pancreatitis in ADPKD patients.
Case presentationHerein, we present a case of a 12-year-old Caucasian girl with recurrent pancreatitis with no identifiable traumatic, metabolic, infectious, drug, or immunologic causes. Structural anomalies of the pancreas, including cysts, were ruled out by imaging. However, bilateral cystic kidneys were found as an incidental finding. Her family history was negative for pancreatitis, but positive for polycystic kidney disease. Molecular analysis of ADPKD-causing mutations revealed a novel c.9659C>A (p.Ser3220*) mutation in the PKD1 gene confirming the clinical suspicion of ADPKD. Although CP may rarely occur as an extrarenal manifestation of ADPKD with pancreatic cysts, it is unusual in their absence. Thus, molecular analysis of pancreatitis susceptibility genes was performed and a homozygous pathologic c.180C>T (p.G60=) variant of the CTRC gene, known to increase the risk of CP, was confirmed.
ConclusionThis is the first reported case of a pediatric patient with coincidence of genetically determined CP and ADPKD. Occurrence of pancreatitis in children with ADPKD without pancreatic cysts warrants further investigation of CP causing mutations.
En niños, la pancreatitis crónica (CP, por sus siglas en inglés) generalmente se asocia con anomalías anatómicas del páncreas y el tracto biliar, o está genéticamente determinada. La enfermedad renal poliquística autosómica dominante (ADPKD, por sus siglas en inglés) puede presentarse con la formación de quistes extrarrenales, que a veces afecta al páncreas. Suficientemente grandes, estos quistes pueden causar pancreatitis en pacientes con ADPKD.
Presentación del casoPresentamos el caso de una niña caucásica de 12 años con pancreatitis recurrente sin causas identificables traumáticas, metabólicas, infecciosas, farmacológicas o inmunológicas. Las anomalías estructurales del páncreas, incluidos los quistes, se descartaron mediante imágenes. Sin embargo, los riñones quísticos bilaterales se encontraron como un hallazgo accidental. Su historia familiar fue negativa para la pancreatitis, pero positiva para la enfermedad renal poliquística. El análisis molecular de las mutaciones causantes de ADPKD reveló una nueva mutación c.9659C>A (p.Ser3220*) en el gen PKD1 que confirma la sospecha clínica de ADPKD. Aunque la CP rara vez ocurre como una manifestación extrarrenal de ADPKD con quistes pancreáticos es inusual. Por lo tanto, se realizó el análisis molecular de los genes de susceptibilidad a pancreatitis y se confirmó una variante homocigótica patológica c.180C>T (p.G60=) del gen CTRC, que se sabe que aumenta el riesgo de CP.
ConclusiónEste es el primer caso reportado de un paciente pediátrico con coincidencia de CP y ADPKD genéticamente determinados. La aparición de pancreatitis en niños con ADPKD sin quistes pancreáticos justifica una mayor investigación de CP que causan mutaciones.
While alcohol abuse and gallstones are the most common triggers of chronic pancreatitis (CP) in adults, structural anomalies of the pancreas and biliary tract, as well as genetic factors, have to be considered in CP in children. The estimated incidence of CP in young adults is 0.5:100,000 per year and the rare, genetically determined CP represents only a part of it.1 A growing number of genes has been associated with increased risk of CP development.2–4
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent genetic kidney disorder that affects approximately 1:1000 individuals worldwide. Progressive bilateral kidney cyst enlargement leads to end-stage renal disease in 50% of patients.5 Liver cysts, colonic diverticula, or intracranial aneurysms may lead to extrarenal manifestations. Cysts in the pancreas develop in approximately 9% of ADPKD patients. They are usually smaller than 10mm and therefore rarely cause pancreatitis.6,7
In this study, we introduce a case of a girl with CP and concomitant polycystic kidney disease, both genetically determined.
Case descriptionThe now 12-year-old Caucasian girl was initially diagnosed with the first attack of acute pancreatitis at the age of 8. She was treated with bowel rest, intravenous hydration and antibiotics. As pancreatitis is unusual at this age, a search for predisposing anatomical anomalies was initiated. Magnetic resonance scans raised suspicion of pancreas divisum with no intervention needed, as she was without complaints for the following 2 years. However, at the age of 10, she experienced another attack of pancreatitis. Laboratory tests showed normal findings in hematologic tests, but biochemical parameters supported the diagnosis of pancreatitis: pancreatic amylase 3.84ukat/l (N: ≤0.58ukat/l), amylase 4.42ukat/l (N: ≤1.70ukat/l), lipase 108.62ukat/l (N: 0.67–2.34ukat/l), calcium 2.23mmol/l (N: 2.25–2.85mmol/l), C-reactive protein 24mg/l (N: 0–10mg/l), amylase and pancreatic amylase in urine were elevated more than 10 times above the upper normal values. On ultrasonography, the pancreas increased in echogenicity and size (from 120mm at admission to 150mm on day 3). Neither necrosis, abscesses, pseudocysts, nor cysts were detected. However, the pancreatic parenchyma displayed a coarse tissue structure, indicating chronic pancreatitis. The previously suspected pancreas divisum was ruled out by magnetic resonance cholangiopancreatography (MRCP). As a coincidental finding, numerous bilateral cortical kidney cysts up to 11mm large were found. Interestingly, the family history was negative for any gastrointestinal symptoms or pancreatitis, but it was positive for polycystic kidney disease on the maternal side of the pedigree (Fig. 1). The clinical diagnosis ADPKD was stated based on ultrasound diagnostic criteria.8
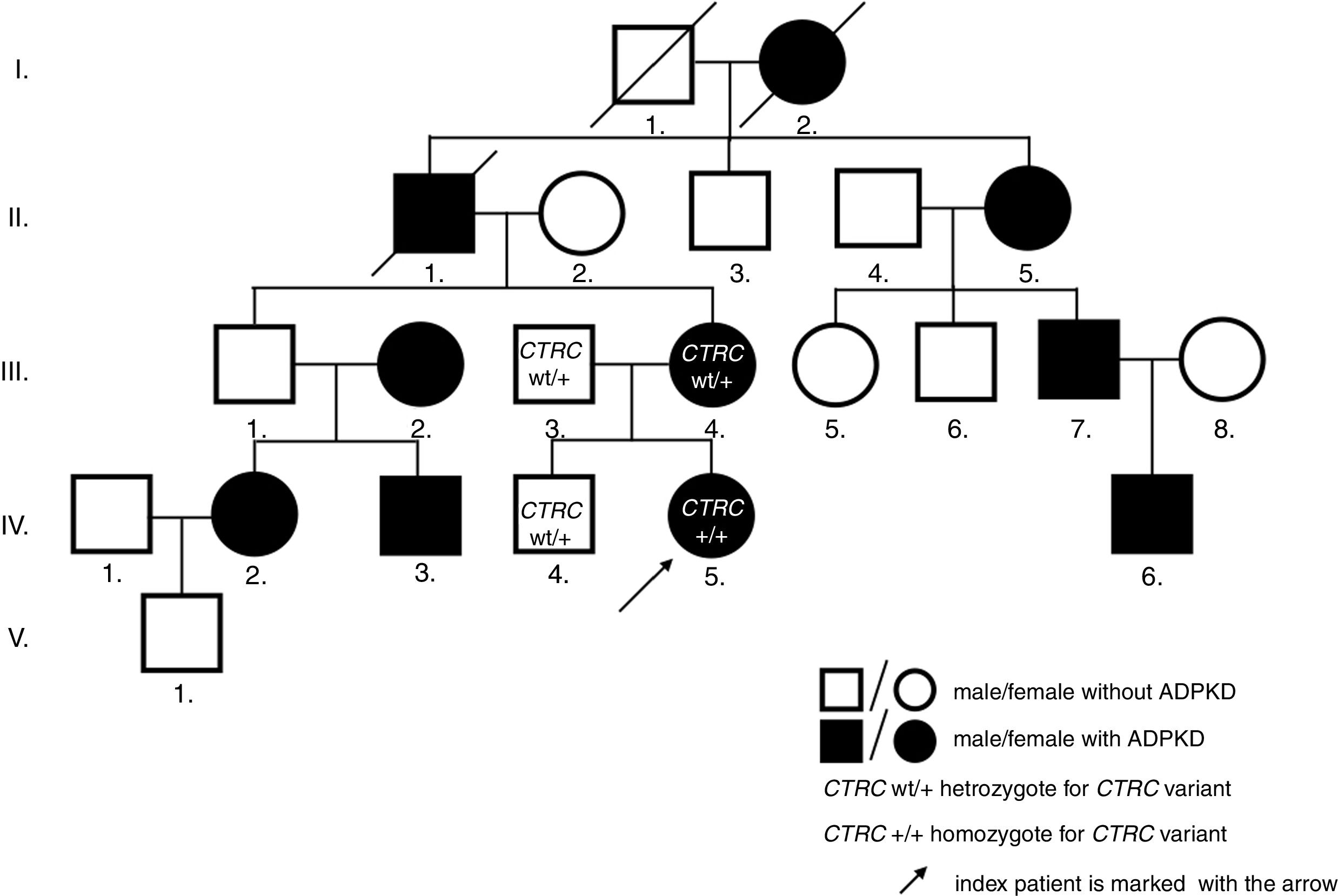
As pancreatic cysts were excluded, ADPKD was found to be unlikely the cause of CP. By this time, other structural anomalies of the pancreas and biliary tree, infectious, toxic, metabolic, or immunological agents were also ruled out. Hence, mutation screening of 4 genes associated with idiopathic CP was performed. In PRSS1, SPINK1, and CFTR, no mutation was found, but the homozygous c.180C>T (p.G60=) variant in the CTRC gene was identified. Both parents as well as her brother were heterozygous carriers.
The genetic confirmation of clinically diagnosed ADPKD followed by linkage analysis, revealing a link to the PKD1 gene. Direct sequencing of its coding sequence identified a novel pathologic nonsense variant c.9659C>A (p.Ser3220*) in our index patient, her mother, and mother's relatives with a clinical picture of ADPKD. The nucleotide replacement creates premature termination codon, thus predicts to truncate the protein, leading to ADPKD phenotype. Renal function assessment in our patient confirmed physiologic glomerular filtration rate, adequate concentration capacity, and blood pressure.
A genetic cause of both, CP and polycystic kidney disease could be identified in the same patient. Currently, the girl is doing well, avoiding fatty food and paying attention to sufficient fluid intake.
DiscussionWe report a rare case of a child with chronic pancreatitis and the incidental finding of numerous renal cysts, confirmed to be due to ADPKD. After excluding pancreatic cysts as a possible cause of CP in ADPKD, we found a homozygous CTRC variant p.G60=predisposing to CP.
To date, only few sporadic cases of concomitant pancreatitis and ADPKD have been described in the literature. It is usually restricted to patients with pancreatic duct compression due to pancreatic cyst development that is causally related to ADPKD.6
A case, presented by Malka et al., described a 38-year-old female patient with ADPKD, abdominal pain, and biochemical parameters consistent with pancreatitis. A single pancreatic cyst, 15mm large, compressing the main pancreatic duct, was concluded to be the cause of the chronic obstructive pancreatitis – an extrarenal manifestation of ADPKD.9 An unusual combination of pancreatic cysts and pancreas divisum in a male patient with ADPKD and two consecutive attacks of acute pancreatitis was seen by Basar et al. Surprisingly, both anomalies appeared for the first time when the patient was 63 years old.10 Multiple pancreatic cysts led to obstructive pancreatitis also in a 47-year-old ADPKD male patient reported by Sastre López et al.11 Most recently, two unrelated cases of recurrent attacks of acute pancreatitis were described in ADPKD individuals with only mild kidney disease; while imaging studies revealed pancreatic cysts in a 38-year-old male described by Yazdanpanah et al., no pancreatic cysts were detected, in a 70-year-old female patient reported by Thyagarajan et al., in whom an enlarged cystic liver was recognized to be the underlying condition of pancreatic duct obstruction.12,13 The only pediatric case of pancreatitis due to multiple pancreatic cysts within ADPKD was reported by Kahn and Palmert in 1996. Compression of the main pancreatic duct in its mid-part and mild dilation in the tail area was revealed in a girl as young as 9 years. Pancreatic enzyme release secondary to duct compression was suspected to be the cause of the pancreatitis.14 This was a unique case, as ADPKD-related pancreatic cysts are rare in children. According to autopsy and imaging studies, these are related to ageing with the maximum prevalence after the third decade.6,7 In summary, the proportion of ADPKD patients who develop pancreatic cysts is small and usually restricted to adult patients. Even among ADPKD patients with pancreatic cysts occurrence of pancreatitis seems to be rare6,7 and the few cases of ADPKD and pancreatitis are clearly defined by the presence of pancreatic cysts with critical pancreatic duct compression.9–12,14
Genetic susceptibility plays an important role in the pathogenesis of CP and genetic alterations in the PRSS1, SPINK1, CFTR, CTRC, CPA1, CEL, CLDN2 and CTRB1-CTRB2 locus may be associated with chronic pancreatitis.2–4 The presence of ADPKD and genetically determined CP may be a coincidence. However, CP is a complex genetic disorder that develops through the interaction of environmental and genetic factors.2 Thus, it cannot be excluded, that changes in CP susceptibility genes together with mutations in ADPKD-causing genes could potentially trigger early-onset pancreatitis. This is supported by our described case with a synonymous c.180C>T (p.G60=) variant.
According to Beer et al. and Paliwal et al. the heterozygous c.180C>T (p.G60=) carriers have an increased risk for CP 1.65-fold and 2.5-fold respectively, whereas homozygotes 8.44-fold and tenfold respectively.15,16 It exerts its effect via a loss-of-function mechanism as disease-related CTRC variants are connected to decreased mRNA levels.15
The association between the variant and the CP was further supported by the study of LaRusch et al. who observed that the presence of the variant increases the risk of CP specifically in individuals with a known genetic background (disease-causing mutations in the genes SPINK1, CFTR, CTRC, and PRSS1) and alcohol/smoking-related CP etiology.17CTRC gene is therefore considered to be a disease modifier gene with pathogenic variants representing one of the most important factors increasing susceptibility to pancreatic damage through the trypsin-dependent pathway.2
The increased risk of pediatric CP linked to the c.180C>T (p.G60=) variant in the CTRC gene was confirmed in a large pediatric Polish cohort of 136 children (with either idiopathic CP or CP related to other genetic/non-genetic risk factors). Heterozygous carriers had 2.8-fold increased risk of CP and homozygotes 23-fold compared to controls. The homozygous c.180C>T (p.G60=) genotype was found to be the strongest CP risk factor among all identified variants in the CTRC gene.18
Whether coincidental combination of ADPKD causing PKD1, PKD2, or GANAB mutations5 and genetic susceptibility to CP has an adverse impact on pancreatic cyst development and/or early pancreatitis manifestation need to be reliably determine by further studies.
CP might occur also independently from ADPKD, and the case of our patient could be a unique coincidence of both diseases at the same time. Further follow-up of the propositus will be important for better understanding of the behavior and natural course of both diseases appearing together. Also, genetic counseling for the family will inform them about the risk of development of one or both disorders in future generations. From a clinical perspective, we suggest that in children with both ADPKD and CP not related to the presence of pancreatic cysts, genetic causes of CP should be considered.
ConclusionTo the best of our knowledge, this is the first case of a pediatric patient with chronic pancreatitis and coincidental autosomal dominant polycystic kidney disease with molecular evidence of ADPKD causing mutation and genetic propensity to CP. Based on our case, we recommend to include genetic factors contributing to development of pancreatic inflammation in the differential diagnosis of pancreatitis in ADPKD. Further observations and analyses are needed to determine whether mutations causing ADPKD are likely to trigger pancreatitis manifestation in patients with genetic susceptibility to CP.
Compliance with ethical standardsAll procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Submission declaration and verificationThe work described here has been approved by all authors, it has not been published previously (except for an abstract) and it is not under consideration for publication in other journal.
Conflict of interestNone.
This work was supported by the Ministry of Health of the Slovak Republic under the project registration no. 2012/5-UKBA-5 and by the Slovak Research and Development Agency under the contract no. APVV-14-0234. We would like to thank Ľudmila Vavrová, PhD. for technical support and helpful discussions.



