
Aunque el fósforo es un elemento indispensable para la vida, en la naturaleza no se encuentra en estado nativo sino combinado en forma de fosfatos inorgánicos (PO43−), con niveles plasmáticos estrechamente regulados que se asocian a efectos deletéreos y mortalidad cuando estos se encuentran fuera de la normalidad. El interés creciente sobre el acúmulo de PO43− en la fisiopatología humana se originó en el papel que se le atribuyó en la patogenia del hiperparatiroidismo secundario a la enfermedad renal crónica. En este artículo revisamos los mecanismos por los cuales se justificaba dicho efecto y conmemoramos la importante contribución de un grupo español liderado por el Dr. M. Rodríguez, ahora hace justo 25 años, cuando demostraron por primera vez el efecto directo del PO43− sobre la regulación de la síntesis y secreción de hormona paratiroidea (manteniendo la integridad estructural de las glándulas paratiroides en su nuevo modelo experimental. Además de demostrar la importancia del ácido araquidónico (AA) y la vía de la fosfolipasa A2-AA como mediadora de respuestas en la glándula paratiroidea, estos hallazgos fueron predecesores de la reciente descripción del importante papel del PO43− sobre la actividad del receptor-sensor de calcio y alimentaron asimismo diversas líneas de investigación sobre la importancia de la sobrecarga de PO43−, no solo en la fisiopatología del hiperparatiroidismo secundario sino también en su papel patogénico sistémico.
Although phosphorus is an essential element for life, it is not found in nature in its native state but rather combined in the form of inorganic phosphates (PO43−), with tightly regulated plasma levels that are associated with deleterious effects and mortality when these are out of bounds. The growing interest in the accumulation of PO43− in human pathophysiology originated in its attributed role in the pathogenesis of secondary hyperparathyroidism in chronic kidney disease. In this article, we review the mechanisms by which this effect was justified and we commemorate the important contribution of a Spanish group led by Dr. M. Rodríguez, just 25 years ago, when they first demonstrated the direct effect of PO43− on the regulation of the synthesis and secretion of parathyroid hormone by maintaining the structural integrity of the parathyroid glands in their original experimental model. In addition to demonstrating the importance of arachidonic acid (AA) and the phospholipase A2-AA pathway as a mediator of parathyroid gland response, these findings were predecessors of the recent description of the important role of PO43− on the activity of the calcium sensor-receptor, and also fueled various lines of research on the importance of PO43− overload not only for the pathophysiology of secondary hyperparathyroidism but also of its systemic pathogenic role.
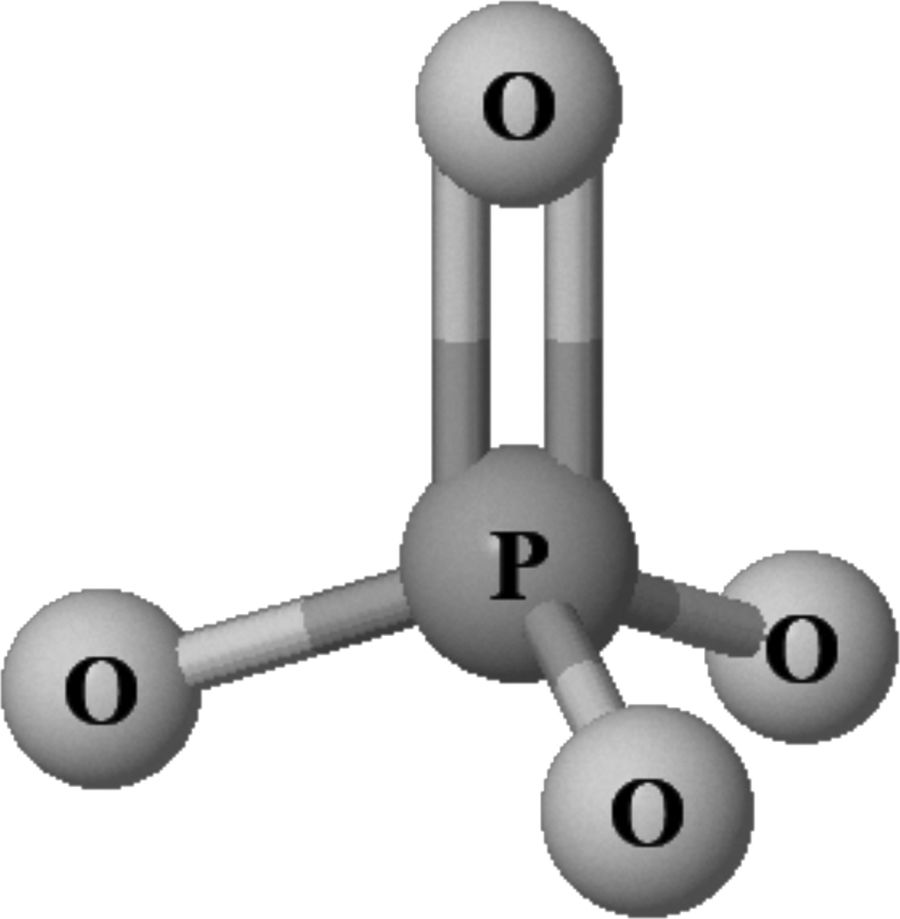
El fósforo (P), (en griego phos phorus o portador de luz), es un elemento indispensable (número atómico 15, peso molecular 30,9 u) que se encuentra en todos los organismos vivos. En estos tiene una doble función: a) estructural, en los ácidos nucleicos (ADN y ARN), las membranas celulares (fosfolípidos) y la fase mineral del hueso (junto al calcio constituye los cristales de hidroxiapatita); b) reguladora, constituyendo el principal intermediario energético en procesos celulares como el metabolismo y activación de proteínas (fosforilación), participando en la curva de disociación del oxígeno de la hemoglobina (2,3-difosfoglicerato) y en procesos de señalización celular (segundos mensajeros como el AMP y GMP cíclicos). En la naturaleza, incluidos los seres vivos, el fósforo no se encuentra en estado nativo, sino combinado principalmente en forma de diversos fosfatos inorgánicos (PO43−) (fig. 1), no inflamables, siendo esta la forma en que medimos los niveles de fósforo en el organismo. Su importancia biológica determina que los niveles de PO43− deban estar estrictamente controlados dentro de un rango fisiológico óptimo y que fuera de este se asocie con efectos deletéreos. De este modo, múltiples estudios epidemiológicos muestran que no solo su carencia es causa de patología, sino que también niveles de PO43− elevados (incluso en rangos de normalidad) se han asociado de forma independiente con enfermedad cardiovascular y mortalidad en la población general y, especialmente, en pacientes con enfermedad renal crónica (ERC), ya sea en diálisis o no1-7.
: compuesto por un átomo de fósforo y átomos de oxígeno en forma tetraédrica. Realizada con el JSME Molecular Editor (http://biomodel.uah.es/en/DIY/JSME/draw.en.htm).")
Si consideramos que los riñones tienen un papel fundamental en la regulación del PO43–, las alteraciones de su homeostasis en la ERC son esperables debido a que el balance depende de su ingesta y excreción. El acúmulo intra- y extracelular de PO43− tiene un rol esencial no solo en la patogenia del hiperparatiroidismo secundario (HPTS) y del complejo chronic kidney disease-mineral and bone disorders (CKD-MBD) sino que además, directa o indirectamente, se puede considerar como un factor de riesgo (no clásico) de morbimortalidad cardiovascular e incluso infecciosa, a través de diversos mecanismos como la inflamación, el estrés oxidativo, las calcificaciones vasculares y valvulares o la fibrosis miocárdica, así como también parece contribuir a la disfunción celular del sistema inmune, a la arterioesclerosis-ateromatosis e incluso al envejecimiento acelerado en los pacientes con ERC1,8-10. Recientemente ha tomado también relevancia el papel del PO43− y los microcristales de fosfato cálcico en la luz tubular renal no solo como efecto secundario sino también como factor causal de la propia progresión de la ERC11,12.
No obstante, el interés creciente sobre el acúmulo de PO43− en la fisiopatología humana se originó en el papel que se le atribuyó en la mencionada patogenia del HPTS. Por ello, el objetivo de este artículo es revisar los mecanismos por los cuales se justificaba dicho efecto y conmemorar la importante contribución de un grupo español a la primera demostración en estudios experimentales del efecto directo del PO43− sobre la regulación de la síntesis y secreción de hormona paratiroidea (PTH) en las glándulas paratiroides, ahora hace justo 25 años13.
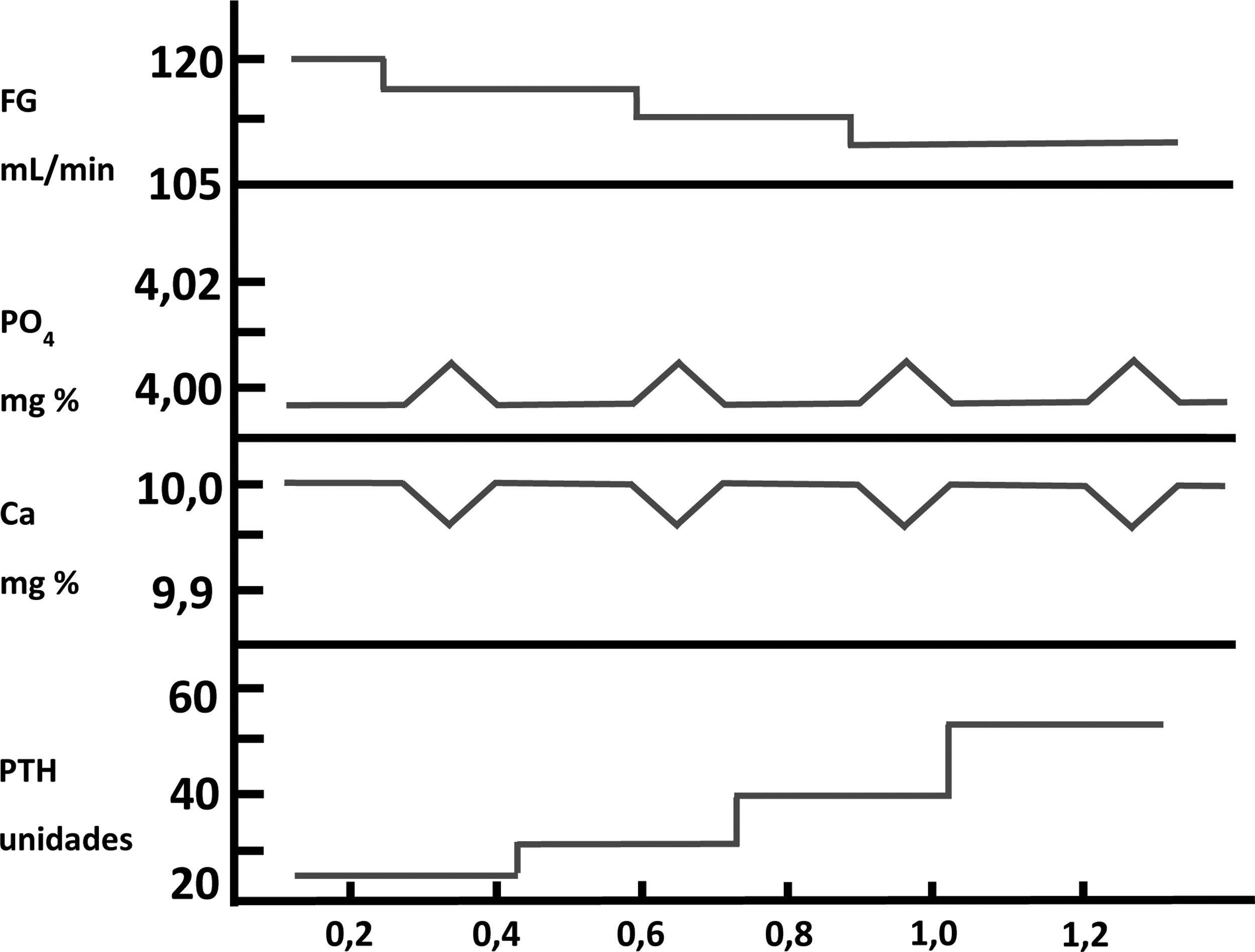
El fósforo en la patogenia del hiperparatiroidismo secundario: mecanismos indirectosDurante muchos años ha existido un gran debate, casi bipolar, sobre si la retención de PO43− o la disminución de la síntesis de calcitriol (1,25-[OH]2-vitamina D), presentes ambos en los pacientes con ERC, eran los factores iniciadores o más importantes en la patogenia del HPTS14-16. Hace ahora exactamente cinco décadas (bodas de oro), Slatopolsky et al.14 observaron un incremento significativo de la PTH en un modelo experimental en perros con ERC alimentados con una dieta alta en PO43−. La interpretación de estos hallazgos fue que elevaciones transitorias de PO43− posprandiales disminuirían el calcio ionizado en sangre, y esta disminución de calcio sería la que estimularía la secreción de PTH. El aumento de PTH no solo normalizaría los niveles de calcio, sino que también disminuiría la reabsorción tubular de fósforo, con el consiguiente incremento de la fosfaturia, y así se normalizarían los valores séricos de ambos iones a expensas de la elevación de la PTH sérica. Ulteriores disminuciones del filtrado glomerular (FG), conducirían a un aumento progresivo de los valores de PTH necesarios para normalizar las cifras de calcio y PO43− en sangre. Estas observaciones constituyeron la «trade-off hypothesis» («hipótesis del trueque» o elevación de PTH –y sus efectos deletéreos consecuentes– a expensas de intentar mantener la homeostasis del calcio y PO43−)17,18 (fig. 2). Estos mismos autores demostraron en otro estudio experimental que la reducción proporcional en la ingesta de PO43− ajustada a la disminución gradual del FG fue capaz de prevenir el HPTS, evidenciado por la presencia de niveles plasmáticos normales de calcio, PO43–, PTH y reabsorción tubular de fósforo en comparación con los controles19. En este modelo experimental, la explicación a las observaciones realizadas se fundamentaron en que no era necesaria la adaptación de las nefronas remanentes al estadio de ERC, en el que dichas nefronas deben responder a una mayor fracción de excreción renal de PO43− con una disminución de su reabsorción tubular (como suele suceder en la ingesta no ajustada de PO43− u otros elementos)20. Esta hipótesis fue postulada por Bricker bajo la denominada «hipótesis de la nefrona intacta»20 y los hallazgos sugirieron entonces que una ingesta constante de PO43− ante una población de nefronas disminuida (ERC) jugaría un papel importante en la patogenia del HPTS20,21.
.")
Patogénesis del hiperparatiroidismo secundario en la enfermedad renal crónica. Representación de la «trade-off hypothesis». Adaptada de Bricker et al.18).
Aunque en el estudio mencionado19 la reducción en la ingesta de PO43− previno el HPTS, no podía descartarse la posibilidad de la contribución de una síntesis mayor de calcitriol inducida por la dieta baja en PO43−. Este hecho sería especialmente relevante sobre todo en etapas tempranas del daño renal, tal y como fue demostrado por Portale et al.15 años más tarde en niños con ERC moderada. El PO43− es un conocido inhibidor de la 1α-hidroxilasa (CYP27B1) y en estos niños se evidenció que una dieta alta en PO43− estimulaba la PTH y disminuía los niveles de calcitriol, mientras que una dieta baja en PO43− estimulaba el calcitriol con descenso secundario de la PTH. Por otra parte, es frecuente que una dieta baja en PO43− se acompañe de un incremento del calcio sérico derivado de la mayor absorción intestinal de calcio secundaria al aumento del calcitriol, de forma que este aumento en el calcio sérico podría explicar al menos en parte el descenso de la PTH. De este modo, Llach y Massry16 demostraron en cuatro pacientes con ERC moderada que la restricción de PO43− interfería con la acción y producción de calcitriol, modificando no solo la absorción intestinal de calcio sino también la respuesta calcémica a la PTH, sugiriendo así un papel importante de la alteración del metabolismo de la vitamina D en la patogenia del HPTS en la ERC.
La entonces denominada disminución de la respuesta calcémica o resistencia esquelética a la acción de la PTH (endógena y exógena) fue otro mecanismo indirecto descrito por el cual la retención de PO43− y/o la hiperfosfatemia podrían contribuir al origen y progresión del HPTS en la ERC22,23, y fue ampliamente estudiada por Rodríguez et al.24-27. Actualmente denominada «hiporrespuesta» a la PTH28, diversos trabajos han demostrado la importancia relativa de la retención de PO43− en la resistencia a la acción de la PTH, entre otros factores24-30. Tal y como se ha revisado en un artículo reciente en esta revista30, la existencia de hiporrespuesta a la acción de la PTH, a través de diversos mecanismos entre los que destaca la retención de PO43–, demandan una mayor síntesis y secreción de PTH para mantener el equilibrio homeostático mineral.
El fósforo en la patogenia del hiperparatiroidismo secundario: mecanismo directoHasta ahora, los mecanismos descritos en la presente revisión sobre el papel del PO43− en la patogenia del HPTS han sido de tipo indirecto (disminución del calcio iónico, disminución en la síntesis de calcitriol o la hiporrespuesta multifactorial a las acciones de la PTH en la ERC). Es bien conocido que tanto la disminución de la concentración de calcio extracelular como la disminución de los niveles de calcitriol tienen un efecto directo estimulador de la síntesis de PTH y la proliferación de las células paratiroideas, mediado a través de sus respectivos receptores (el receptor sensor de calcio [RSCa] y el de la vitamina D [RVD])31-34. De hecho, Sherwood et al.35 no consiguieron encontrar evidencias de un efecto directo del PO43− en la regulación de la PTH in vivo.
Sin embargo, López-Hilker et al.36, en otro trabajo experimental en perros con ERC sujetos a una restricción de PO43− y calcio para evitar la hipercalcemia, observaron que el nivel sérico de PTH descendió aun sin cambios en el calcio y calcitriol. Estos hallazgos sugerían que el control del HPTS era independiente de los niveles plasmáticos de calcio y calcitriol. Años más tarde, en un modelo experimental en ratas, Yi et al.37, también observaron datos consistentes de la existencia de mecanismos independientes de calcio y calcitriol y, por tanto, de un posible efecto directo del PO43− sobre la función paratiroidea.
Por otra parte, siendo difícil soslayar el efecto directo del calcitriol en la disminución de la síntesis y secreción de PTH cuando se prescribe una dieta baja en PO43–, Kilav et al.38 también demostraron que ratas deficientes en vitamina D de segunda generación, alimentadas con una dieta baja en PO43− y calcio, presentaban hipofosfatemia asociada a niveles bajos de ARNm de PTH en ausencia de hipercalcemia o aumento de niveles de calcitriol, sugiriendo un efecto no transcripcional del PO43–, a diferencia de los efectos directos del calcitriol disminuyendo la transcripción del gen de la pre-pro-PTH. Posteriormente se ha demostrado que este efecto no transcripcional del PO43− es en realidad postranscripcional, implicando la unión de proteínas transactivantes (proteínas que actúan en trans o adenosine-uridine-rich binding factor [AUF1]) a dominios cis (elemento cis) ubicados en la región 3’ no traducida del ARNm de la PTH, orquestadas por la isomerasa Pin1, induciendo en último término un aumento de la estabilidad del ARNm de PTH39-42.
Finalmente, Hernández et al.43 también habían descrito en un trabajo preliminar los efectos de una dieta alta en PO43− en un modelo de rata in vivo y encontraron, respecto a las ratas que ingirieron una dieta estándar en PO43–, un incremento significativo de los niveles de PTH asociado a un rápido incremento de la expresión del ARNm de PTH sin afectar los niveles de calcio o calcitriol. Como demostraron posteriormente, esta observación no se acompañaba de modificaciones en la expresión del RVD o del RSCa43-45, sugiriendo que una carga oral de PO43− podría estimular directamente la síntesis de PTH y que no era mediada por una menor expresión de los receptores de calcio o calcitriol.
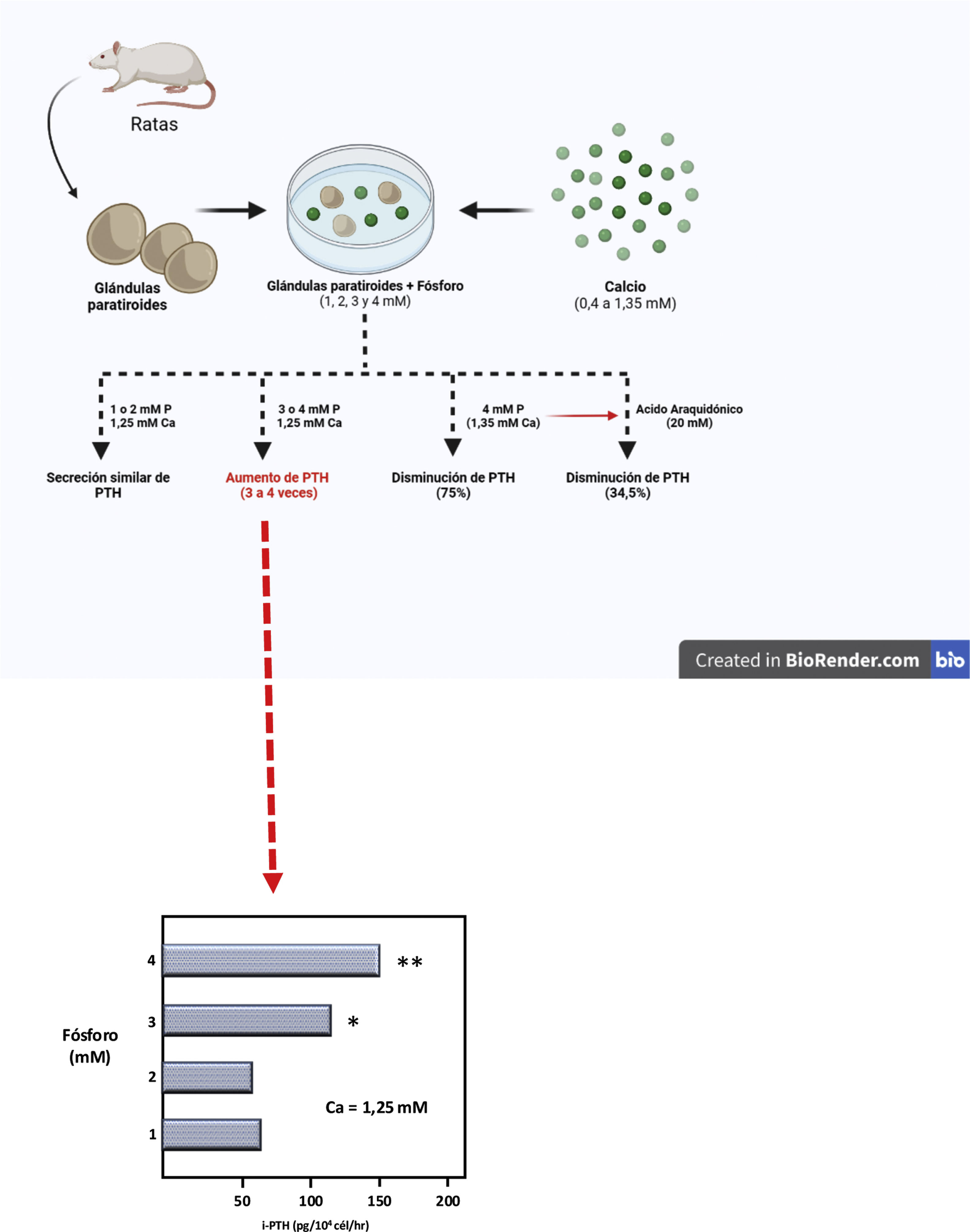
De este modo y empleando un modelo experimental completamente original, basado en el mantenimiento de la integridad estructural de la glándula paratiroidea, un grupo español liderado por el doctor M. Rodríguez describió por primera vez in vitro y de un modo indudable el efecto directo del PO43− sobre la secreción de PTH (IX Congreso Latinoamericano de Nefrología en San Juan de Puerto Rico, octubre 20-23, [1994] y el Xlllth International Congress of Nephrology en Madrid, julio 2-6, [1995]). Así, Almadén et al.13 publicaron en 1996 su trabajo en el que usaron glándulas paratiroides íntegras frescas de rata que fueron incubadas con diferentes concentraciones de PO43− (1,2,3, y 4mM) y subsecuentemente fueron expuestas a diferentes concentraciones de calcio en un rango de 0,4 a 1,35mM. Los autores observaron que, con una concentración de calcio de 1,25mM, la secreción de PTH fue similar con las concentraciones de PO43− de 1 y 2mM; sin embargo, concentraciones de PO43− de 3 y 4mM produjeron un aumento en la secreción de PTH tres a cuatro veces mayor, respectivamente, en comparación al PO43− 1mM (fig. 3). Asimismo, mientras que las concentraciones de 1 o 2mM de PO43− con incremento en la concentración de calcio de 0,6 a 1,35mM redujeron la secreción de PTH al 37%, en PO43− 4mM el mismo aumento de la concentración de calcio solo inhibió la secreción de PTH al 75% (fig. 3). Esta demostración pionera del rol esencial de la integridad estructural de la glándula paratiroidea y de mantener los contactos célula-célula constituyó un punto de partida fundamental para acelerar el avance del conocimiento del impacto del PO43− en la patogénesis del HPTS en laboratorios de todo el mundo. De hecho hasta entonces, con el mismo objetivo de evaluar el posible efecto directo del PO43− en la función paratiroidea, los estudios in vitro utilizaban células paratiroideas dispersas, con menos respuesta a cambios en el calcio extracelular que las glándulas paratiroides intactas de Almadén et al.13, debido a la disminución de la expresión del RSCa46.
, independiente del calcio (Ca), sobre la estimulación de la secreción de hormona paratiroidea (PTH) en la glándula paratiroidea. Las glándulas fueron incubadas durante 1 hora en concentración de calcio de 1,25mM y concentración variable de fósforo de 1,2,3 y 4mM.")
Representación teórica del modelo usado por Almadén et al.13 para la demostración del efecto directo del fósforo (P), independiente del calcio (Ca), sobre la estimulación de la secreción de hormona paratiroidea (PTH) en la glándula paratiroidea. Las glándulas fueron incubadas durante 1 hora en concentración de calcio de 1,25mM y concentración variable de fósforo de 1,2,3 y 4mM.
En dicho estudio seminal de Almadén et al.13, los autores también abordaron el estudio de los mecanismos de señalización intracelular que mediaban el efecto directo del PO43− sobre la secreción de PTH. Así, la adición de ácido araquidónico (AA, sustrato que inhibe la vía de señal intracelular), al medio de incubación de PO43− 4mM y calcio 1,35mM redujo la secreción de PTH al 34,5%. La conclusión del estudio fue pues que en este modelo que usaba glándulas paratiroideas íntegras frescas de rata, la elevación de PO43−in vitro aumentaba directamente la secreción de PTH actuando a través de la vía de la fosfolipasa A2 (PLA2)-AA.
Posteriormente, también demostraron in vitro que, en glándulas de pacientes con HPTS severo, la secreción de PTH aumentó en respuesta al PO43− 3 y 4mM en comparación a 2mM a pesar de la presencia de una alta concentración de calcio en el medio, lo que se acompañaba de un aumento de los niveles de pre-pro-PTH47. Asimismo, este mismo grupo demostró cómo una elevación aguda de PO43− sérico sin cambios en la concentración de calcio iónico estimuló también la secreción de PTH in vivo48, el efecto de elevadas concentraciones de PO43− en la producción de AA en tejido paratiroideo in vitro49, la regulación de la producción de AA por el calcio intracelular50, evidenciando que la estimulación de la secreción de PTH por niveles de PO43− elevados podía prevenirse aumentando los niveles de calcio intracelular. Asimismo revisaron la importancia del AA y la vía fosfolipasa A (2)-AA como mediadora de respuestas en la glándula paratiroidea51. Además, se demostró que el mantenimiento de niveles elevados de PO43− sérico durante la construcción de curvas calcio-PTH en hemodiálisis (utilizando un líquido de diálisis con alto contenido o libre de PO43−) previno en parte la inhibición de la secreción de PTH por el calcio (normal o elevado)52.
Por otra parte, desde el laboratorio de Slatopolsky et al. (J of Investigative Medicine, abril [1995]), usando el mismo modelo experimental de glándulas paratiroideas intactas de Almadén et al.13, se publicó en este mismo año 1996 el efecto del PO43− en la dieta sobre los niveles de PTH, ARNm de la PTH y la hiperplasia paratiroidea en ratas urémicas y normales53. Los autores observaron que el peso de las glándulas y la PTH sérica fueron similares en ambos grupos expuestos a una dieta baja en PO43− (0,2%), pero hubo un aumento significativo de la PTH sérica, peso y ADN de las glándulas paratiroideas procedentes de las ratas urémicas alimentadas con una dieta alta en PO43− (0,8%) comparadas con ratas urémicas alimentadas con dieta baja en PO43−. Adicionalmente observaron que el efecto estimulador del PO43− extracelular sobre la producción de PTH no se produjo cuando la síntesis de proteínas fue inhibida con cicloheximida, sugiriendo que la acción del PO43− en las células paratiroideas requería de síntesis proteica. Asimismo, la tasa de crecimiento de las células paratiroideas fue independiente de los niveles de calcio y calcitriol. Previamente, Denda et al.54 ya habían demostrado que el efecto del PO43− sobre el crecimiento de las glándulas paratiroideas era muy rápido en ratas urémicas, tras dos meses de la inducción de insuficiencia renal, y que el 90% del crecimiento ocurría durante los primeros tres días, potencialmente atribuido a la participación de protooncogenes como c-fos, c-jun y PRAD-1.
Curiosamente en el mismo año, reconociendo los trabajos de los dos grupos liderados por los doctores Rodríguez y Slatopolsky, y con el mismo objetivo de investigar el efecto directo del PO43− sobre las células paratiroideas in vitro, Kjaerulff et al. del grupo danés del Dr Olgaard55, utilizaron dos tipos de preparaciones de tejido paratiroideo bovino: células paratiroideas dispersas y cortes de tejido paratiroideo, ambas incubadas durante cuatro h en un medio normal (1,0mM) o alto en PO43− (3,5mM) y observaron un incremento significativo de la liberación de PTH en los cortes de tejido paratiroideo incubados en medio alto en PO43− pero no en la preparación con células dispersas, sin observarse cambios en el «set-point» de calcio. El grado de estimulación de la liberación de PTH con medio alto en PO43− fue significativamente mayor comparado con un medio bajo en calcio (0,8mM), un 172% por encima del valor basal (1,0mM de PO43−) y un 139% por encima en un medio con un nivel elevado de calcio (1,8%). Sus resultados también demostraron que el PO43− estimula directamente la liberación de PTH en cortes de glándulas paratiroideas bovinas y no en preparaciones de células dispersas, lo que confirmó que el mantenimiento de una arquitectura normal en las paratiroides era esencial para reproducir el efecto estimulador de la elevación del PO43− en la secreción de PTH.
Efecto del fósforo sobre las glándulas paratiroidesEn condiciones normales, las células paratiroideas están en estado quiescente, raramente entran en mitosis; sin embargo, es bien conocido que en la ERC varios factores como la hipocalcemia y el déficit de calcitriol, inducen el crecimiento de las glándulas paratiroides, estimulando la proliferación de células paratiroideas, inicialmente a expensas de hiperplasia celular y, consecuentemente, la síntesis y secreción de PTH56-59. Ya hemos mencionado que una dieta alta en PO43–, ya sea por mecanismos directos o indirectos, promovía el crecimiento de las paratiroides desde etapas tempranas tal y como lo demostraron Denda et al. en estudios experimentales en ratas54. Por el contrario, también se demostró que una dieta baja en PO43− y la administración de calcitriol prevenían la hiperplasia inducida por la uremia y la secreción de PTH. Así, Dusso et al.60 demostraron en ratas urémicas que una dieta baja en PO43− fue capaz de prevenir la hiperplasia de las glándulas paratiroides a través de un aumento de la proteína y el ARNm de p21 en tejido paratiroideo. La proteína codificada por el gen p21 es un inhibidor de las kinasas dependientes de ciclinas (complejos Cdk) y, por lo tanto, un regulador del ciclo celular. Asimismo, se demostró en este estudio que una dieta alta en PO43− indujo la expresión del factor de crecimiento transformante-α (TGF-α) como señal autocrina que estimulaba el crecimiento de las paratiroides. Tales elevaciones de TGF-α en la glándula paratiroidea inducidas por dietas hiperfosfatemiantes y la consecuente activación del receptor del factor de crecimiento epidérmico (EGFR por su acrónimo inglés) fueron identificadas como el factor determinante de las disminuciones del RVD y el origen de la resistencia al control del HPTS con el calcitriol o sus análogos con el avance de la ERC61. Es importante también resaltar que, a medida que progresa la ERC, no solo disminuye la expresión del RVD sino también la expresión del RSCa y del receptor del factor de crecimiento fibroblástico 23 (FGF23-FGFR) durante su evolución desde la hiperplasia difusa a hiperplasia nodular59. Más recientemente, también se demostró que una alta demanda de secreción de PTH, promovida bien por una dieta muy rica en PO43− o pobre en calcio, inducía diferentes patrones de hiperplasia paratiroidea en ausencia de uremia, una situación que podría ser importante en estadios precoces de ERC62.
Es también interesante destacar en este punto que la regulación de la PTH por el PO43− implica también ciertos micro-ARN (miARN)63,64. Los miARN son ARN pequeños no codificantes con funciones vitales en la homeostasis y el desarrollo del organismo y sabemos que la enzima Dicer está involucrada en la etapa final del procesamiento de los miARN. En este sentido, por ejemplo, hemos aprendido recientemente que los ratones knockout específicos de Dicer en las células paratiroides (PT-Dicer -/-) tienen niveles séricos normales de PTH, pero no pueden aumentar la PTH en hipocalcemia o insuficiencia renal, a diferencia de los controles64. También, además de modular la secreción de PTH, los miARN son esenciales para mantener intactas las glándulas paratiroides. Los ratones sin gen Dicer no expresan miARN en las células paratiroideas y pierden sus glándulas paratiroides después del nacimiento. Esto indica que los miARN no son esenciales para el desarrollo embrionario de las glándulas paratiroides, sino más bien para su integridad durante el período posnatal. En ausencia de glándulas paratiroides, los ratones adultos PT-Dicer -/-, una fuente adicional de PTH, distinta de las células tiroideas o del timo, contribuye al mantenimiento de la concentración sérica normal de PTH, pero no puede ser estimulada por hipocalcemia o estado urémico64.
Otros aspectos relacionados con la sobrecarga de fósforo en la enfermedad renal crónicaNo es el propósito de este artículo revisar toda la compleja fisiopatología del HPTS sino, como se ha mencionado, conmemorar los 25 años del importante descubrimiento del efecto directo del PO43− sobre la glándula paratiroides, en la que tuvieron un papel tan relevante investigadores españoles13,53. Sin embargo, sí es necesario mencionar la importancia del descubrimiento de una fostatonina, el factor de crecimiento fibroblástico 23 (FGF23)65,66, una hormona producida por los osteocitos y cuya producción es estimulada principalmente por la sobrecarga de PO43–, entre otros factores67,68. Hoy sabemos que su elevación se produce desde estadios precoces de la ERC, incluso antes de la elevación de la PTH sérica, la cual también estimula a su vez la producción de FGF2369-71. Como fosfatonina, el FGF23 tiene una acción no solo fosfatúrica al inhibir la expresión de los canales Na-Pi 2a y 2c a nivel tubular renal (disminuyendo la reabsorción tubular de PO43− y aumentando así su excreción urinaria) sino que también inhibe la 1α-hidroxilasa (CYP27B1) renal (responsable de la síntesis de calcitriol) como estimula la 24-hidroxilasa (CYP24A1) (aumentando su catabolismo)69,70. Este nuevo mecanismo de contrarregulación de la sobrecarga de PO43− nos permite revisitar la ya antigua, pero aún vigente, hipótesis del «trade-off’»17,21,70,72 y la importancia del PO43− en la patogenia del HPTS en la ERC, al aportar un nuevo mediador anteriormente desconocido. Este incremento de FGF23, a su vez, se explica al menos en parte asociado a la menor expresión de su correceptor Klotho, necesario para ejercer su acción en los tejidos diana (paratiroideo, vascular, cerebral, tubular renal) demostrados en varios estudios y que, en el caso del tejido renal, produciría a su vez resistencia a la acción del FGF2373. Como ya es conocido ahora, la acción del FGF23 está mediada normalmente por una afinidad canónica entre su receptor FGFR y su correceptor Klotho10,74, de modo que al disminuir la producción de Klotho y/o la expresión del FGFR como consecuencia de la ERC73-76, se producirá otra hiporrespuesta hormonal (resistencia) que se une a la de la PTH u otras hormonas como la insulina o la hormona del crecimiento30. Esta hiporrespuesta al FGF23 conducirá a un aumento adicional de los niveles de FGF23 necesarios para ejercer su acción adaptativa fosfatúrica en presencia de ERC y/o sobrecarga de fósforo, pero con efectos deletéreos secundarios (como otro «trade-off», en este caso a expensas de FGF23)68,73,77-79. De hecho, tanto la disminución de Klotho como el aumento de FGF23 propios de la ERC han sido claramente asociados con el envejecimiento acelerado y la desproporcionadamente alta mortalidad de los pacientes con ERC, especialmente en sus estadios avanzados o en diálisis2,10,80, convirtiendo la ERC en un desafortunado modelo experimental humano de senescencia8,9. De este modo, la retención intra- y extracelular de PO43− constituye uno de los estímulos principales para la síntesis y secreción de ambas hormonas (PTH y FGF23), ya sea de forma directa e indirecta, con el fin de aumentar la fosfaturia, entre otros efectos. Por otra parte, PTH y FGF23 actúan de manera competitiva en la regulación enzimática de la producción y catabolismo de la vitamina D. Esta interrelación hormonal compleja se exacerba con la disminución progresiva del parénquima renal al progresar la ERC. Adicionalmente, la reducción secundaria de klotho, el aumento de PO43− y el incremento de FGF23, actúan como estímulos proinflamatorios, siendo elementos clave en el estado inflamatorio consustancial a la ERC e involucrado en múltiples efectos deletéreos asociados a la pérdida de la función renal (desarrollo de anemia y resistencia a la acción de los agentes estimulantes de la eritropoyesis, aparición de un síndrome de malnutrición proteico-energética, disfunción endotelial), así como a las calcificaciones y aterosclerosis precoces y aceleradas81-83.
CorolarioEn condiciones normales, la homeostasis del PO43− extracelular está coordinada entre la absorción intestinal, la excreción renal, así como su entrada y salida del hueso84. Tanto las glándulas paratiroides como el hueso detectan el aumento de PO43− extracelular respondiendo a través de un incremento en los niveles de PTH y FGF23, respectivamente, con el propósito de aumentar la fosfaturia; sin embargo, el mecanismo molecular intrínseco es desconocido a diferencia de lo que ocurre con las interacciones del calcio o los calcimiméticos con el RSCa, la vitamina D con el RVD e incluso con el FGF23 y su propio receptor33,46,59,75. Durante muchos años se ha intentado encontrar el receptor del PO43− en la glándula paratiroides para intentar explicar su efecto directo, postulándose incluso la posibilidad de la existencia de un canal transportador.
No ha sido hasta recientemente que se ha demostrado la importancia del RSCa en este papel. Este receptor se encuentra en la superficie de las membranas de múltiples células y pertenece a la familia de proteínas denominadas receptores acoplados a proteínas G. Ya era sabido también que los calcimiméticos podían superar el efecto estimulador de la elevación de PO43− sobre la secreción de PTH in vitro e in vivo85. Recientemente, Geng et al.86 usaron una técnica de cristalografía de rayos X, para estudiar la estructura tridimensional del dominio exterior del RSCa en estado activo y en reposo. Se observó que los iones de calcio son los principales activadores del RSCa pero requiere de la fijación de aminoácidos en su forma activa. Asimismo, se detectó que el dominio extracelular del RSCa tenía cuatro puntos de fijación de aniones multivalentes ocupados por el PO43− y SO42− y que los iones PO43− mantenían estable la forma inactiva, promoviendo así la secreción de PTH. Por otra parte, Centeno et al.87, recientemente mostraron en modelos experimentales murinos que el aumento de PO43− en concentraciones fisiopatológicas para la ERC inhibe la actividad del RSCa de forma antagónica no competitiva. De este modo, estos resultados nos muestran finalmente que el RSCa es un sensor también del PO43–, explicando el mecanismo intrínseco por el que el PO43− estimula directamente la secreción de PTH y proporciona un mecanismo por el cual las concentraciones elevadas de PO43− pueden ejercer efectos directos en los tejidos que expresan el RSCa.
En resumen, la sobrecarga de PO43− conduce a la activación de diversos mecanismos, directos e indirectos, encaminados a mantener su homeostasis88. La presencia de ERC no hace sino crear al menos dos círculos viciosos en lo que al complejo CKD-MBD se refiere (aumento de PTH y de FGF23), con sus efectos deletéreos sistémicos (doble «trade-off» que no solo afecta al hueso sino también al sistema cardiovascular)1,8,68, en un intento de normalizar el metabolismo mineral. Con la progresión de la ERC, la acumulación de PO43− (y por ende la importancia de la restricción de las fuentes de PO43− fundamentalmente inorgánico para prevenir el desarrollo de HPTS), ya sea a través del estímulo del FGF23, la inhibición de la síntesis de calcitriol, la hipocalcemia resultante, su efecto negativo sobre la respuesta calcémica a la acción de la PTH o la estabilización de la forma inactiva del RSCa, claramente bloquea todos los mecanismos conocidos de contrarregulación diseñados para mantener la homeostasis del metabolismo mineral en situaciones de salud. Además, esta acumulación de PO43− contribuye directamente a producir efectos claramente nocivos mediados por múltiples mecanismos que inciden sobre el sistema cardiovascular o el propio riñón1-8,12,89-92 y que parecen explicar la importantísima asociación de la sobrecarga de PO43− sobre la morbimortalidad, especialmente en pacientes con ERC.
Conflicto de interesesJB ha recibido honorarios como consultor, ponente o ayudas de viaje de Amgen, Abbvie, Sanofi y Vifor-Fresenius-Renal Pharma. PU ha recibido honorarios como consultor o ponente de Amgen, Astellas, GSK, Hemotech, Leo-Pharma, Sanofi y Vifor-Fresenius-Renal Pharma. AT ha recibido honorarios como consultor de Alnylan Pharm. JFNG ha recibido honorarios como consultor, ponente o ayudas de viaje de Abbvie, Amgen, Sanofi-Genzyme, Shire y Vifor-Pharma. JF ha recibido honorarios como consultor de Vifor-Pharma.
Algunos autores pertenecen a la Red de Investigación Renal (REDINREN RD 16/0009/0022) y RICORS2040 (RD21/0005/0013), Instituto de Salud Carlos III. Asimismo agradecemos al Sr. Ricard Pellejero su inestimable ayuda bibliográfica.



