

La enfermedad de Fabry o también llamada de Anderson-Fabry (EF) es una enfermedad rara, causada por variantes patogénicas en el gen GLA, localizado en el cromosoma X. Este gen interviene en el metabolismo de los glucoesfingolípidos y variantes patogénicas en el mismo causan déficit o ausencia de la α-galactosidasa A, ocasionando el depósito de globotriaosilceramida en todo el organismo. Las mujeres presentan una expresión fenotípica variable y de mejor pronóstico que los varones. Esto es debido al fenómeno de inactivación del cromosoma X. Presentamos un caso clínico de enfermedad de Fabry en una mujer con afectación predominantemente renal y demostramos cómo el fenómeno de la inactivación del cromosoma X es tejido dependiente, mostrando una inactivación preferencial del alelo mutado a nivel renal.
Fabry disease or also called Anderson–Fabry disease (FD) is a rare disease caused by pathogenic variants in the GLA gene, located on the X chromosome. This gene is involved in the metabolism of glycosphingolipids and its pathogenic variants cause a deficit or absence of α-galactosidase A causing the deposition of globotriaosylceramide throughout the body. Females have a variable phenotypic expression and a better prognosis than males. This is due to the X chromosome inactivation phenomenon. We present a clinical case of Fabry disease in a female with predominantly renal involvement and demonstrate how the X chromosome inactivation phenomenon is tissue dependent, showing preferential inactivation of the mutated allele at the renal level.
La enfermedad de Fabry (EF) es una enfermedad de depósito lisosomal ligada al cromosoma X, causada por mutaciones en el gen GLA que conducen a una actividad deficiente o ausente de la enzima α-galactosidasa A (α-Gal A). Esto produce una acumulación de glucoesfingolípidos como globotriaosilceramida (Gb3) y su forma deacilada, globotriaosilesfingosina (lyso-Gb3), en plasma, orina y en los lisosomas y citosol de varios tipos de células (endoteliales vasculares, podocitos, cardiomiocitos, células del músculo liso arterial etc.). El gen GLA se localiza en Xq22.1 y actualmente hay descritas más de 900 variantes patogénicas distintas en este gen, la mayoría de tipo missense1–3. La incidencia de la EF clásica es de entre 1:40.000 y 1:117.000 varones nacidos vivos y de 1:3.200 si se incluyen los fenotipos de inicio tardío1–4.
El acúmulo de Gb3 y lyso-Gb3 está relacionado con la proliferación de células de músculo liso, la remodelación vascular, el estrés oxidativo, la activación de la inmunidad innata y la liberación de citocinas proinflamatorias1,5. El fenotipo de la enfermedad depende de la actividad enzimática residual. Un nivel ausente o < 1% de α-Gal A ocasiona el «Fabry clásico», de inicio pediátrico y afectación multiorgánica, mientras que una actividad enzimática residual da lugar a las formas de inicio tardío.
Las manifestaciones clínicas del Fabry clásico se presentan en el sistema nervioso en forma de dolor neuropático crónico y crisis de dolor intenso («crisis de Fabry»), parestesias en edades tempranas y en adultos se puede presentar en forma de accidentes isquémicos transitorios o accidentes cerebrovasculares isquémicos. Es frecuente la presencia de angioqueratomas, hipohidrosis e intolerancia al calor y dismorfismo facial. A nivel gastrointestinal puede haber dolor abdominal, diarrea o estreñimiento. En las formas clásicas es casi constante la córnea verticillata1–6, siendo al igual que la mayoría de los síntomas, excepto los cardiacos mucho menos frecuente en las formas tardías. La afectación renal y cardiaca (hipertrofia ventricular, fibrosis endomiocárdica, arritmias) se presentan en edades más avanzadas.
La afectación renal puede ir desde microalbuminuria, proteinuria hasta desarrollo de insuficiencia renal progresiva y necesidad de tratamiento renal sustitutivo. La podocituria es la manifestación más temprana que se produce a nivel renal, precediendo el desarrollo de proteinuria1,3,4,6.
La variabilidad en los niveles de la actividad residual de la enzima α-Gal A se correlacionan con el fenotipo que puede ser desde asintomático a grave en las formas clásicas1,5,6. Las formas de inicio tardío muestran un fenotipo preferentemente cardiaco.
El diagnóstico de esta enfermedad suele ser complejo y tardío, con edad media de diagnóstico de 23 años en varones y 32 en mujeres2. Se debe sospechar ante la evidencia clínica y/o antecedentes familiares. En los varones la determinación de actividad α-Gal A es suficiente para llegar al diagnóstico, mientras que en mujeres es necesario el estudio genético, ya que pueden tener actividad de α-Gal A dentro del rango de normalidad3,4,6. La evidencia de acumulación de Gb3 lisosomal en biopsias renales o cardiacas, aunque invasiva, puede ser necesaria cuando las manifestaciones clínicas o la interpretación de la variante en el gen GLA no son concluyentes4. Biomarcadores como el Gb3 urinario y el Gb3 y lyso-Gb3 en sangre pueden ser útiles para el diagnóstico y, más dudosamente, para la monitorización del tratamiento5.
Durante muchos años, las mujeres con EF fueron consideradas como solo portadoras de la enfermedad, pero se ha evidenciado que pueden presentar un espectro clínico amplio, desde casos asintomáticos hasta casos graves similares a los de los varones, pese a la actividad enzimática conservada1. La explicación más plausible de la variabilidad fenotípica en las mujeres es la inactivación del cromosoma X (ICX) o lionización, un proceso mediante el cual las mujeres igualan la dosis de genes con respecto a los varones XY mediante la inactivación de uno de sus cromosomas X en cada célula1-4,6,7. Otros factores como genes modificadores o factores epigenéticos o ambientales también podrían contribuir a la expresión variable de la enfermedad en mujeres.
Exponemos un caso clínico de EF con afectación predominantemente renal y mínima expresión clínica a otros niveles. Demostramos la correlación clínica con la ICX a nivel renal.
Descripción del casoMujer de 35 años diagnosticada a los 17 años tras hallazgo de enfermedad de Fabry en progenitor (padre) con variante patogénica en el exón 7 del gen GLA (NM_000169.3): c.1102delinsTTATAC, p. (Ala368Leufs*25). Se trata de una paciente sin antecedentes de interés, con clínica de dolores ocasionales en extremidades inferiores y dishidrosis en ambas manos de larga evolución. No presentaba angioqueratomas ni otras alteraciones cutáneas. En el momento del estudio destaca la presencia de proteinuria de 500 mg/24 h, sin microhematuria, función renal normal y estudio de función cardiaca con ecocardiografía sin alteraciones. Los niveles de α-Gal en plasma eran de 70%. Fue valorada por oftalmología con hallazgo de córnea verticillata. Con estos hallazgos se decide ingreso para realización de biopsia renal, dado el grado importante de proteinuria en una mujer joven con EF, con el objetivo de descartar otras cusas de nefropatía proteinúrica.
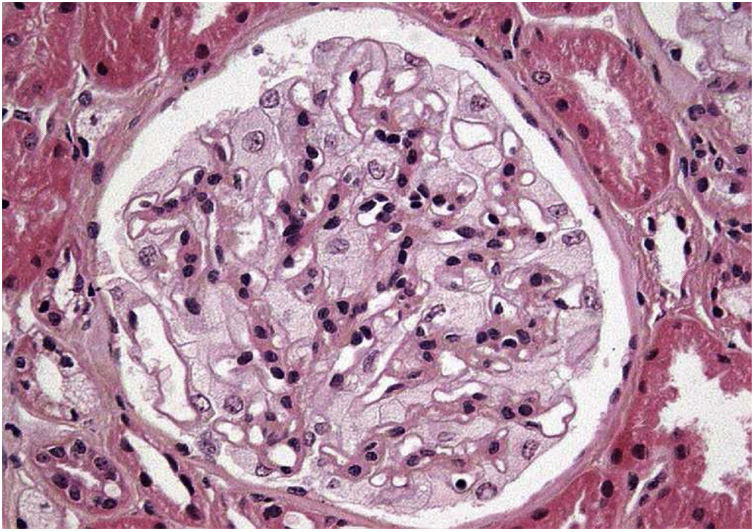
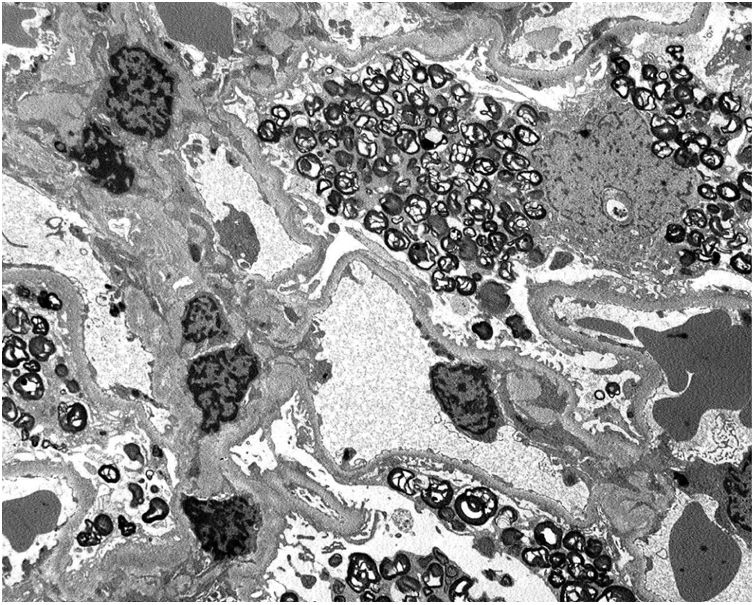
Los resultados de la biopsia renal muestran (fig. 1) aumento de la matriz mesangial de forma focal y segmentaria con incremento de la celularidad. Tanto las células endoteliales como las epiteliales muestran citoplasma amplio y multivacuolar. También se objetivan células espumosas tubulares. No se identifica afectación intersticial o vascular. En la microscopía electrónica (fig. 2) se observa una masiva cantidad de inclusiones de aspecto mielínico predominantemente localizadas en el interior de los podocitos, con una mínima proporción de dichas inclusiones en células endoteliales, mesangiales y tubulares. Se diagnostica con los resultados de EF y se decide inicio de tratamiento de reemplazo enzimático (TRE) con agalsidasa beta e inhibidores de la enzima convertidora de angiotensina (IECA).


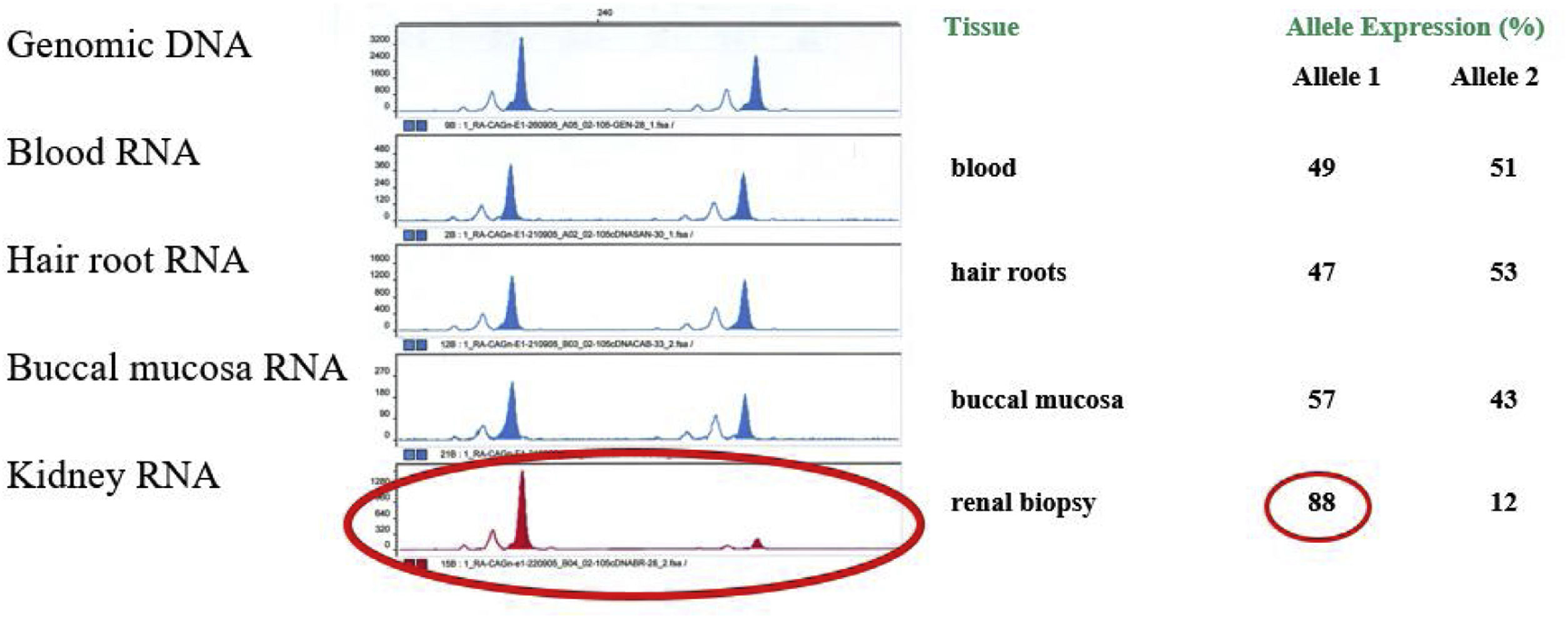
Dada la marcada afectación renal con escasa afectación de otros órganos (salvo córnea verticillata), se realizó un estudio del nivel de ICX en distintos tejidos de la paciente: sangre periférica, pelo, mucosa bucal y riñón. Para ello, se analizó el patrón de ICX mediante análisis transcripcional del del polimorfismo CAG del gen del receptor de andrógenos, utilizando la metodología reportada previamente8. Se observó un nivel de expresión similar de ambos alelos del cromosoma X (mutado y salvaje) en todos los tejidos analizados, salvo en el riñón, que presentaba una expresión significativamente mayor del alelo mutado (fig. 3).
 en distintos tejidos de la paciente. En el primer panel se muestra la amplificación del gen AR en el ADN genómico realizada para corregir la amplificación preferencial del alelo de menor tamaño (alelo 1). En el resto de los paneles se muestra la expresión de ambos los alelos del gen AR se muestran en sangre, raíces capilares, mucosa bucal y riñón. La tabla de la derecha muestra la cuantificación de los niveles de expresión relativos de ambos alelos AR que muestran una inactivación aleatoria del cromosoma X en todas las muestras analizadas, excepto en el riñón (> % del alelo mutado).")
Determinación del estado de inactivación del cromosoma X (ICX) en distintos tejidos de la paciente. En el primer panel se muestra la amplificación del gen AR en el ADN genómico realizada para corregir la amplificación preferencial del alelo de menor tamaño (alelo 1). En el resto de los paneles se muestra la expresión de ambos los alelos del gen AR se muestran en sangre, raíces capilares, mucosa bucal y riñón. La tabla de la derecha muestra la cuantificación de los niveles de expresión relativos de ambos alelos AR que muestran una inactivación aleatoria del cromosoma X en todas las muestras analizadas, excepto en el riñón (> % del alelo mutado).
La paciente continúa en TRE desde hace 18 años. Durante el seguimiento se mantiene estable desde el punto de vista renal, con filtrados glomerulares superiores a 90 mL/min/1,73 m2 (CKD-EPI) y proteinuria oscilante entre 300 y 800 mg/24 h. Sigue controles en cardiología sin desarrollo de alteraciones de la conducción ni de la función cardiaca, objetivándose en últimas pruebas de imagen cardiaca valor anormalmente reducido del T1 nativo en el segmento medio inferolateral del ventrículo izquierdo, compatible con depósito graso lisosomal miocárdico atribuible a la EF.
DiscusiónLas manifestaciones clínicas de la EF en mujeres se presentan en torno a 70% de los casos con una alta variabilidad fenotípica; esto se debe mayoritariamente a la ICX1. Las enfermedades de herencia ligada al cromosoma X, como la enfermedad de Fabry, generalmente se manifiestan de forma más grave en los varones que en las mujeres. Esto es debido a que los varones son hemicigotos para los genes del cromosoma X (solo tienen una copia), mientras que las mujeres al disponer de dos cromosomas X mantienen actividad enzimática. En las mujeres se produce el fenómeno de la inactivación del cromosoma X durante la primera semana del desarrollo embrionario. El X inactivo puede ser el paterno o el materno y se inactiva de forma aleatoria, pero permanente. Normalmente la asignación aleatoria de la ICX hace que se inactive 50% de los cromosomas X de origen materno y 50% de los de origen paterno, resultando en un mosaico de células que expresan cada una uno de los dos cromosomas X. Sin embargo, en un porcentaje bajo de mujeres se dará lugar una ICX sesgada con > 75% de expresión de uno de los dos X; y en un porcentaje mucho menor (5%) una ICX extremadamente sesgada > 90%1,4,7,9-12. Los porcentajes de ICX sesgado varían dependiendo de la edad y el tipo de tejido, estimándose alrededor de 5-14% en las recién nacidas, 14% en las mujeres de ≤ 25 años, 16-37% en mujeres mayores de 60 años y de 49% en las centenarias1,9. Los tejidos descendientes de cada una de estas células embrionarias mantienen el mismo patrón de ICX1,13.
En la EF, 50% de expresión de la enzima funcional sería teóricamente suficiente para no presentar síntomas graves de la enfermedad, ya que entre las células que expresan el cromosoma X salvaje o mutado hay una cooperación metabólica con aporte de enzima a las células mutadas1,9,14. También alternativamente las células deficitarias de la enzima podrían dividirse menos eficientemente, siendo eventualmente reemplazadas por las no deficitarias, lo que se denomina selección celular11,14.
En el caso de la EF, la α-Gal A es captada por los receptores de manosa-6-fosfato en la membrana plasmática para entrar por endocitosis en las células deficientes, sin embargo, algunos estudios sugieren que la cooperación metabólica es pobre en esta enfermedad (secreción insuficiente de α-Gal A, defecto de glicosilación de la enzima, absorción deficiente) y que la mayoría de las pacientes con EF y ICX aleatoria desarrollarán síntomas crecientes con la edad9,10,14.
En varios estudios como el de Řeboun et al., realizado con 35 mujeres con EF (26 con ICX aleatorio y nueve con ICX sesgado), o el Echevarria et al., con 56 mujeres (30% con ICX sesgado), se observa una correlación entre el patrón de ICX y la actividad de α-Gal A9,12. En el reciente estudio de Řeboun et al. no se detectó relación entre el tipo de variante y la actividad α-Gal A en mujeres (hallándose pacientes con variantes truncantes o missense graves y con una alta actividad de la enzima). Se observó también el efecto del envejecimiento sobre el patrón de ICX con cambios en dos de ocho pacientes a los 6-10 años12. Los datos sobre el impacto de ICX en la EF no son uniformes en todos los estudios y todavía falta evidencia.
La concordancia de ICX en los distintos tejidos ha sido estudiada en la población general con resultados variables y escasísimos estudios hasta el momento en EF. El estudio de Echevarria et al. estudió el patrón de ICX en muestras de sangre periférica, mucosa bucal, piel y epitelio urinario en muestra de orina. Observó un patrón de ICX sesgado en 30% de los casos y observó que el patrón de ICX en sangre se correlacionaba con el del resto de muestras de forma estadísticamente significativa9. Sin embargo, en el presente estudio, la ICX sesgada detectada en riñón no se observa en el resto de los tejidos, correlacionándose con la afectación principalmente renal en la paciente. Asimismo, en la cohorte de Řeboun et al. se objetivaron discordancias en los patrones de ICX en las distintas muestras de seis de 15 pacientes (40%)12. Esto es compatible con la discordancia de ICX entre las diferentes células madre pluripotenciales, lo que lleva a la afectación, incluso grave, de determinados órganos con menor afectación de otros.
El proceso de ICX todavía está lejos de ser conocido completamente, pero parece claro que se trata de un fenómeno dinámico y complejo. Esta complejidad incluye una proporción de genes que escapan de la ICX, diferencias específicas de tejido y de célula a célula del proceso de ICX, y el intrincado papel específico de genes y región génica de la metilación del ADN ejercida tanto en los cromosomas X activos como en los inactivos X. Se ha determinado un papel regulador importante a nivel transcripcional del X mediado por el gen XIST (X inactive specific transcript)10,15,16, así como a nivel de la traducción en proteínas o por mecanismos epigenéticos (metilación del DNA, modificación de histonas, etc.)15–17. La inactivación comienza en el centro de inactivación del cromosoma X (XIC), donde el ARN XIST no traducido se acumula y recubre el futuro cromosoma X inactivado, seguido de cambios epigenéticos a nivel de todo el cromosoma. En las células somáticas, el cromosoma X inactivado es visible como el cuerpo de Barr18.
Aunque cierto sesgo en la inactivación del X podría atribuirse a variantes en el gen XIST (implicadas en casos familiares de ICX sesgado), tales variantes ocurren solo en raras ocasiones14, considerándose la selección celular y el sesgo aleatorio (dada la pequeña cantidad de células presentes en el momento de la implantación) como las causas más frecuentes de ICX sesgada1,11,14. Todavía hay muchas preguntas sin resolver, en particular con respecto al momento de inicio y reversión de ICX, la secuencia de eventos moleculares y la red reguladora de XIST y del resto de mecanismos responsables15.
La mayoría de los estudios que investigan el papel de la metilación del ADN en la EF analizan el gen HUMARA (human androgen receptor), ya que es rápido y económico19. Sin embargo, es importante tener en cuenta que el estudio del gen HUMARA no puede distinguir entre los dos cromosomas y, por lo tanto, no puede predecir la gravedad de la enfermedad en una mujer con ICX muy sesgada. Además, los enfoques clásicos para medir la ICX sesgada podrían no ser suficientes para explicar las manifestaciones de la enfermedad en las mujeres. La metilación del ADN alelo específico en la región promotora del gen GLA también puede influir en los niveles de expresión del alelo mutado, con implicaciones en la gravedad de la EF. Por lo tanto, los enfoques que distinguen entre el alelo mutado y no mutado al analizar la metilación del ADN en el promotor GLA pueden ser mucho más informativos. En general, el estudio de la ICX no es algo que se realice en la práctica clínica ni hay suficiente evidencia para recomendarlo. A pesar de esto, se ha llegado a incluir como criterio de inicio de tratamiento específico para la EF4.
El presente caso demuestra la ausencia de correlación de la ICX en sangre periférica con lo que ocurre en otros órganos. Aunque no se propone como una práctica clínica habitual, resulta interesante desde el punto de vista fisiopatológico.
FinanciaciónNinguna.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses..



