



Passenger lymphocyte syndrome (PLS) causes immune-mediated hemolysis in solid and bone marrow transplant recipients. Donor-derived antibodies against the recipient erythrocyte drive the pathogenesis. It is a rare entity in kidney transplantation, and most of the cases are self-limited.
Case presentationA 36-year-old woman presented with fatigue 13 days after living donor renal transplantation. The operation was uneventful, and she was discharged with normal graft functions on the 11th day of transplantation Findings were consistent with cold agglutinin disease at her admission. However, the cold agglutinin test was negative. Eventually, she was diagnosed with PLS. Refractory intravascular hemolysis and frank hemoglobinuria were also present in the patient. Hemolysis was resistant to steroids, intravenous immunoglobulin (IVIG), and Rituximab. Because of life-threatening anemia related to refractory PLS, mycophenolate and tacrolimus were interrupted. However, hemolysis persisted. Following that, immunoadsorption (IA) treatment was obtained. Unfortunately, graft loss occurred due to rejection despite the resolution of PLS after IA.
ConclusionPLS is a rare and usually self-limited entity. Our case was an atypical refractory PLS that resembled cold agglutinin disease. Also, frank hemoglobinuria was observed related to severe intravascular hemolysis. These features have not been described before in PLS, to the best of our knowledge. Additionally, IA treatment had never been reported in the literature for PLS, as far as we know. Treatment and management could be a challenge in refractory PLS. Rituximab, IVIG, and extracorporeal treatments could be beneficial. It should be borne in mind that refractory PLS can cause graft and patient loss.
El síndrome de linfocitos pasajeros (PLS) causa hemólisis inmunomediada en receptores de trasplantes sólidos y de médula ósea. Los anticuerpos derivados del donante contra el eritrocito receptor impulsan la patogénesis. Es una entidad rara en el trasplante de riñón y la mayoría de los casos son autolimitados.
Presentación del casoUna mujer de 36 años presentó fatiga 13 días después del trasplante renal de donante vivo. La operación transcurrió sin incidentes y fue dada de alta con las funciones normales del injerto el día 11 del trasplante. Los hallazgos coincidían con la enfermedad por crioaglutininas en el momento de su ingreso. Sin embargo, la prueba de crioaglutininas fue negativa. Finalmente, le diagnosticaron PLS. La paciente también presentó hemólisis intravascular refractaria y hemoglobinuria franca. La hemólisis fue resistente a los esteroides, la inmunoglobulina intravenosa (IgIV) y el rituximab. Debido a la anemia potencialmente mortal relacionada con PLS refractario, se interrumpieron el micofenolato y el tacrolimus. Sin embargo, persistió la hemólisis. A continuación, se obtuvo el tratamiento de inmunoadsorción (IA). Desafortunadamente, la pérdida del injerto ocurrió debido al rechazo a pesar de la resolución de PLS después de la IA.
ConclusiónEl PLS es una entidad rara y generalmente autolimitada. Nuestro caso fue un PLS refractario atípico que se asemejaba a la enfermedad por crioaglutininas. Además, se observó hemoglobinuria franca relacionada con hemólisis intravascular grave. Estas características no se han descrito antes en PLS, según nuestro leal saber y entender. Además, el tratamiento IA nunca se había informado en la literatura para PLS, hasta donde sabemos. El tratamiento y el manejo podrían ser un desafío en PLS refractarios. El rituximab, la IgIV y los tratamientos extracorpóreos podrían ser beneficiosos. Debe tenerse en cuenta que los PLS refractarios pueden provocar la pérdida del injerto y del paciente.
Passenger lymphocyte syndrome (PLS) is a rare disease that occurs after solid or bone marrow transplantations at various rates.1 Antibodies produced by B lymphocytes and plasma cells of the donor lead to immune hemolytic anemia that is usually self-limited. Treatment is based on the transfusion of red blood cells with donor blood type. Additional immunosuppressants such as Rituximab and plasmapheresis may be required in refractory cases.1 We used immunoadsorption (Immusorba TR-350) to treat refractory passenger lymphocyte syndrome. Hemolysis was resolved after six sessions of immunoadsorption. Unfortunately, mixed-type allograft rejection occurred following the resolution of hemolysis. Refractory PLS is a rare entity after renal transplantation. The best treatment modality in these cases is a manner of debate. Immunoadsorption might be a therapeutic option in refractory PLS patients.
Case presentationA 36-year-old female patient was admitted to the clinic with fatigue on the 13th day of renal transplantation. She developed an end-stage renal disease related to seronegative pauci-immune crescentic glomerulonephritis. Then, preemptive transplantation was performed from a 0/6 human-leukocyte-antigen (HLA) matched 62-year-old-male living-unrelated donor. Donor-specific antibodies against HLA were negative. Recipient and donor blood types were A Rh-positive and 0 Rh-positive, respectively. Although panel reactive antibodies were negative, the patient history was remarkable for two pregnancies (G2P2A0) and the previous transfusion of two units of red blood cells. Given the circumstances (0/6 HLA match, history of pregnancy and transfusion, old donor and young recipient age), 2mg/kg/day for three days of Anti-thymocyte globulin (ATG) was administered for induction therapy. She was discharged without complication on the 11th day of transplantation with a serum creatinine level of 0.8mg/dL. Hemoglobin level was 7.2g/dL, and serum LDH level was within the normal range at discharge. The maintenance treatment included tacrolimus, mycophenolate mofetil, and prednisolone.
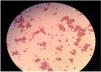
Physical examination revealed jaundice and pallor. The patient had sinus tachycardia at a rate of 120 beats per minute. Laboratory data showed that hemoglobin: 5.0g/dL (Reference (Ref): 12.5–16g/dL), hematocrit: 9.8% (Ref: 37.5–48%), platelet: 165,000/mm3 (Ref: 155000–375000/mm3), reticulocyte count: 5%, serum lactate dehydrogenase: 1840U/L (Ref: 135–250U/L), ferritin: 221ng/mL (Ref: 13–150ng/mL), folate: 6.6ng/mL (Ref: 3.1–17.5ng/mL), Vitamin B12: 364pg/mL (Ref: 191–663pg/mL), serum creatinine: 1.6mg/dL (Ref: 0.7–1.4mg/dL), and serum haptoglobin: 3mg/dL. (Ref: >30mg/dL) Blood smear showed agglutinated erythrocyte clusters and spherocytes resembling cold agglutinin disease (Fig. 1). Two units of A Rh-positive red blood cells were transfused because of severe hemolytic anemia. Direct antiglobulin test before transfusion was positive for both C3 and IgG (IgG (3+), C3 (2+), and IgM antibodies could not be evaluated). Also, indirect antiglobulin and cold agglutinin tests were negative. No sign of bleeding was present in the computed tomography. Cytomegalovirus, Epstein–Barr virus, and Parvovirus DNA tests were negative. However, anti-A1 antibodies were measured by gel agglutinin assays (Coombs kart, Across DiaPro A.S, Gebze/Turkey) were positive for immunoglobulin G and M with 1/32 and 1/16 dilution, respectively. These serological findings were consistent with Passenger lymphocyte syndrome (PLS). Hence, methylprednisolone (1mg/kg/day following 1000 mg IV methylprednisolone) and intravenous immunoglobulin (1 g/kg in two days) were started. Also, tacrolimus and mycophenolate mofetil were discontinued due to life-threatening PLS (Table 1).

On the fourth day of admission, hemolysis persisted. (LDH increased to 3852U/L) Hence, immunoadsorption (Immusorba TR-350) following Rituximab (375mg/m2 for one day) was administered (Table 1). However, serum creatinine levels reached 2.6mg/dL with hemoglobinuria during these therapies. (Fig. 2A, B and 3) Donor-specific HLA antibodies (DSA) were not detected. It was presumed as acute tubular injury related to hemoglobin cast nephropathy, and allograft biopsy could not be performed due to life-threatening anemia.
Immunosuppression of the patient during the clinical course.
| Post-transplant follow-up (day) | Maintenance immunosuppression | Other immunosuppressive | Trough levels of TAC (ng/mL) |
|---|---|---|---|
| 0 | TAC+MMF+Steroid | ATG (Induction therapy) | 6.5 |
| 1 | TAC+MMF+Steroid | ATG (Induction therapy) | 5.4 |
| 2 | TAC+MMF+Steroid | ATG (Induction therapy) | 15.6 |
| From 3 to 10 | TAC+MMF+Steroid | – | Between 10.2 and 15.2 |
| 11 (discharge) | TAC+MMF+Steroid | – | 15.1 |
| 13 (2nd admission) | TAC+MMF+Steroid | – | 18.6 |
| 14 | – | Steroid (1000 mg)+IVIG (Treatment for IHA) | – |
| 15 (PLS diagnosis) | – | Steroid (1 mg/kg/day)+IVIG (Treatment for PLS) | – |
| 16 | – | Steroid (1 mg/kg/day)+Rituximab (Treatment for PLS) | – |
| From 17 to 35 | – | Steroid (1 mg/kg/day)+IA (Treatment for PLS) | – |
| From 36 to 38 | TAC+MMF+Steroid | – | 2.1 |
| 39 (allograft rejection) | TAC+MMF+Steroid | – | 4.2 |
| From 40 to 45 | TAC+MMF+Steroid | ATG+PP+IVIG (Antirejection treatment) | 9.6 |
| From 46 to 57 | TAC+MMF+Steroid | ATG+PP+IVIG (Antirejection treatment) | Between 5.5 and 10.2 |
| 58 | TAC+MMF+Steroid | – | 4.2 |
| 59 | TAC+MMF+Steroid | – | 2.4 |
| 60 (graft loss) | TAC+Steroid | – | 2.3 |
Abbreviations: IVIG; intravenous immunoglobulin, IA; immunoadsorption, IHA; immune hemolytic anemia, MMF; mycophenolate mofetil, PLS; passenger lymphocyte syndrome, PP; plasmapheresis, TAC; tacrolimus.


LDH, serum creatinine, and hemoglobin levels during follow-up. The yellow lightning icons indicate A Rh-positive transfusion. The red lightning icons indicate 0 Rh-positive transfusions. Abbreviations: ATG; anti-thymocyte globulin, IA; immunoadsorption, IVIG; intravenous immunoglobulin, PLS; passenger lymphocyte syndrome, PP; plasmapheresis.
Hemolysis resolved after six sessions of immunoadsorption (IA) during follow-up (Fig. 3). Antibody titers persisted in the first four sessions of IA and then turned negative. Hence, tacrolimus and mycophenolate mofetil were restarted. But, on the 26th day of the second admission, serum creatinine reached 4mg/dL, and donor-specific antibodies against HLA class II were positive (DQ6 7000 MFI with Luminex method, DQB1*0604 2851 MFI with single-antigen bead). Allograft biopsy showed antibody-mediated and Grade-2B cellular rejection according to Banff 2015 classification (Glomerulitis score (g): 1, peritubular capillaritis score (ptc): 1, cg: tubulitis score (t): 3, intimal arteritis score (v): 2, interstitial infiltration score (i): 3), interstitial fibrosis score (ci): 1, tubular atrophy score (ct): 1, transplant glomerulopathy score (cg): 0. No signs of arterial hyalinization or mesangial matrix expansion were present. 1 of 34 glomeruli was global sclerotic). Six sessions of plasmapheresis, methylprednisolone (500mg/d for three days), and ATG (2mg/kg for five days) were administered. Also, intravenous meropenem was started due to bacterial pneumonia on the 43rd day of admission. However, serum creatinine increased to 6.6mg/dL after these therapies. Rebiopsy showed mostly chronic changes. Hence, allograft loss occurred on the 47th day of the second admission (60th day of transplantation), and the patient started hemodialysis thrice weekly.
DiscussionPost-transplant anemia is an overlooked diagnosis but was reported as high as a prevalence of 90% at the early stages of kidney transplantation.2,3 The pathogenesis of post-transplant anemia is multifactorial. It includes allograft dysfunction, iron deficiency, surgical blood loss, drugs, inflammation, erythropoietin deficiency, and hemolysis.2,3 Hemolytic anemia is a life-threatening condition that requires the determination of the underlying cause. Post-transplant hemolytic anemia could be divided into three categories. Autoimmune hemolytic anemia, delayed hemolytic transfusion reaction, and PLS are immune-mediated hemolysis. Also, non-immune-mediated hemolysis (medications, hemoglobinopathies) and thrombotic microangiopathies could cause hemolytic anemia.3
PLS causes immune-mediated hemolysis in solid and bone marrow transplant recipients. Donor-derived antibodies against the recipient erythrocyte are responsible for the pathogenesis.4 The reported incidence varies 8–40%; however, the exact incidence is more than reported due to underdiagnosing.5 A study showed that PLS is the most common cause of hemolysis after transplantation.6
The main effectors are B lymphocytes and plasma cells transplanted with lymphoid tissue during transplantation. These cells produce antibodies against recipient red cell antigens. Besides minor ABO, other antibody mismatches like Rh, Kell, Kidd, and Jka could cause PLS after various solid organ and allogeneic stem cell transplantations.7 The most substantial risk factor is ABO-incompatible kidney transplantation. Most of the cases that have been published after kidney transplantation are related to minor ABO mismatches. Additionally, Rh antibodies (Anti-D) were shown as the cause of PLS in renal transplant recipients.8,9 Both ABO and Rh antibodies generally cause mild hemolysis that starts in the first two weeks of transplantation. They usually cause self-limiting hemolysis up to 8 weeks. However, severe or persistent hemolysis up to 6 months have been reported in both minor ABO and Rh antibodies-related PLS.8,9 PLS does not occur in every case that develops antibodies from the donor lymphocyte. It is unclear what to trigger for the development of PLS. Also, no correlation between the severity of hemolysis and titers of antibodies has been demonstrated.7
Recipient T-lymphocyte inhibition with immunosuppression may allow donor B cell activation and maximize antibody production. Other risk factors are previous sensitization, small bowel or lung transplantation (due to a larger volume of lymphoid tissue). Also, cyclosporine treatment and infections were shown as risk factors in some publications.7,10 In our case, the recipient had A Rh-positive, and the donor had 0 Rh-positive blood group. Also, she had a history of two pregnancies and received induction therapy with ATG.
The diagnosis was made by hemolytic anemia with a positive direct antiglobulin test (DAT). Besides that, donor-derived red cell antibodies against the blood groups of recipient serum confirm the diagnosis. Immunoglobulin G and less often immunoglobulin M or C3 antibodies are detected in DAT.4,7 In our case, PLS was diagnosed after exclusion of cold agglutinin disease. To the best of our knowledge, PLS resembling cold agglutinin disease has not been reported before.
Hemolysis usually occurs between four days to three weeks after transplantation in patients with PLS.7 Also, the clinical presentation ranges from mild hemolysis to fatal anemia with allograft failure. The differential diagnosis includes hemolysis related to drugs, infections, disseminated intravascular coagulation, and thrombotic microangiopathy.3
There is no definitive treatment for PLS. Treatment is supportive and involves transfusions with blood products of the donor blood group. Increasing doses of corticosteroids have been the most used agent, though without good evidence of efficacy. On balance, the potential benefit may outweigh the short-term risks of toxicity. Also, plasmapheresis and Rituximab have been used.1,4,7
Another issue is whether the drugs used in the maintenance immunosuppression that causes anemia should be stopped or not. The physician should make an individual decision by analyzing the harm and benefit for the patient. Also, IA is an effective therapy for the clearance of antibodies before ABO-incompatible solid organ transplantations and pure red cell aplasia.11.
Immunoadsorption protocols are commonly used for the clearance of hemagglutinins before ABO-incompatible kidney transplantations.12,13 IA protocols before transplantation aim to reduce Anti A/B IgG titers below 1/8 before transplantation. Glycosorb, a selective ABO column, is the most commonly used type of immunoadsorption in these patients. Generally, four sessions of Glycosorb IA were shown as sufficient to reduce Anti A/B IgG titers below 1/8 before transplantation.12,13 However, additional sessions might be required at both pre-and post-transplant periods due to a rebound increase in antibody titers. Besides that, IVIG and Rituximab are frequently used in these protocols to prevent rebound of hemagglutinins.12,13 IA has never been used for passenger lymphocyte syndrome so far, to the best of our knowledge.
The mechanism of IA is the high-affinity removal of immunoglobulins by adsorbers. IA could reduce IgG level as an amount of 300% of plasma IgG. This amount is 2 to 3 times greater than therapeutic plasmapheresis.14 Because we could not access Glycosorb to remove Anti-A antibodies selectively, we decided to use Immusorba TR-350 for refractory PLS. Immusorba TR-350 has tryptophan as the ligand, which adsorbs antibodies. It is an effective extracorporeal treatment method to remove both IgG and IgM.15 Hence, we used Immusorba TR-350 to adsorb both Anti-A IgG and IgM antibodies. After six sessions of immunoadsorption, Anti-A IgG antibodies turned negative from 1/32 titer. Hemolysis severity could show no correlation with antibody titers in PLS.7 Following that, our patient had persistent and severe hemolytic anemia despite relatively low titers of Anti-A antibodies. Antibody titers persisted in the first four sessions of IA and then turned negative. Besides that, hemolysis parameters were improved after the initiation of IA. (Figure 3) It could be related to the rebound of hemagglutinins similar to Glycosorb protocols for ABO-incompatible kidney transplantation.12,13 Moreover, it is not strictly possible to exclude the natural decline of antibodies during the disease.
The prognosis of PLS is usually self-limited, and it resolves within three weeks after the initial symptom. Rarely, allograft and patient loss were reported.7,16 Hemoglobin cast nephropathy and acute rejections are the common causes of allograft loss. Death could occur due to complications from transplant surgery or infectious, thrombotic, and immune-mediated complications.7,17 Mycophenolate mofetil and tacrolimus were discontinued due to life-threatening anemia between the 13th and 35th days of transplantation. Rejection and graft loss occurred possibly related to immunosuppressive withdrawal despite other treatments (Intravenous immunoglobulin, high dose corticosteroids, and IA). It could be reasonable not to interrupt the maintenance immunosuppressives as much as possible especially in PLS patients with high immonological risk.
In conclusion, PLS is a rare and usually self-limited entity. However, it should be kept in mind that refractory PLS may cause graft and patient loss. Therefore, careful follow-up of patients for hemolytic anemia should be recommended in patients with minor blood group incompatible solid organ transplants. Rituximab, IVIG, and extracorporeal treatments could be beneficial in refractory PLS.
Informed consentInformed consent was obtained from the patient.
FundingNone.
Conflict of interestNone.
We would like to thank Melek Yanasik for her contribution to the article.



