





INTRODUCTION
Advances in medical genetics in recent decades have allowed for improvements in the diagnosis of familial nephropathies with broad phenotypic variability that until now were referred to as non-specific nephropathies. Overall, these nephropathies can manifest as glomerular disease (active urine sediment with proteinuria and/or haematuria) or as interstitial nephropathy (hyperuricaemia with or without gout, inactive or barely active urine sediment, and often with symptoms of polyuria and polydipsia). However, high variability within families often makes the diagnosis difficult.
Since the discovery of the UMOD gene1 and the recent implementation of its molecular study in cases of familial nephropathy with interstitial clinical findings and an autosomal dominant inheritance pattern, the number of patients identified with mutations in this gene has increased considerably.
We describe the case of a family with a mutation of the UMOD gene that presents with great intra-familial phenotype variability in which genetic study has made early diagnosis of young family members possible. In addition, we review the literature on this entity and the cases described in the literature to date.
CLINICAL CASE
The index case is a male who began being seen in our centre at the age of 19 years due to a six-month history of general malaise and fatigue with alterations in renal function (creatinine 1.8mg/dl). He had a five-year history of hyperuricaemia with no episodes of gout that was controlled with allopurinol (100mg/day). Ultrasound of the kidneys and bladder revealed a decrease in the size of both kidneys (9-10cm) and there was no proteinuria or haematuria in the urine sediment. A renal biopsy (Figure 1) revealed non-specific changes: 4 glomeruli were normal and 3 were sclerotic, the tubules had an atrophic appearance and there was a lymphomononuclear infiltrate. The vessels were not affected and immunofluorescence was negative.
The patient had slowly-progressing renal failure and at 39 years of age he underwent a live-donor kidney transplant (his wife) without incident. The patient is currently 42 years, has very stable renal function with baseline creatinine levels of 1.5mg/dl-1.7mg/dl and is treated with allopurinol 100mg/day.
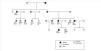
The family history indicates that various members of the family have renal involvement with hyperuricaemia with different levels of renal failures and polyuria with polydipsia (Figure 2) (Table 1). One brother (III-2), with chronic kidney disease and hyperuricaemia, underwent a transplant at 47 years. Another sister (III-1), also with chronic kidney disease and hyperuricaemia (at 35 years of age she has a creatinine level of 1.8mg/dl). The mother (II-4) of the three siblings also has chronic kidney disease, started dialysis at 34 years of age and died at 66 years of age.
The diagnosis of familial juvenile hyperuricaemic nephropathy was made by mutational analysis of the UMOD gene using direct sequencing of the 10 coding exons (exon 2-exon 11) from the genomic DNA of the index case. Mutational analysis allowed for the identification of the c.606G>C (pW202C) sequence variant in heterozygosis (Figure 3). This variant is located in exon 3 of the UMOD gene and has not been described previously in the literature, though another mutation that alters the same amino acid has been described [c.605G>C (p.W202S)2]. The c/606G>C variant (p.W202C) was not identified in more than 200 control chromosomes analysed and it alters the tryptophan 202 amino acid that is conserved in orthologous proteins. In addition, the segregation of this familial variant was studied and it was found to be shared by all of the affected members. Different variant effect predictors (Condel, Sift, Polyphen) predicted that this is a pathogenic mutation. Because of the above, we concluded that the c.606G>C (p.W202C) variant was very likely the causal pathogenic mutation for the disease.
The diagnosis of this pathogenic mutation allowed for pre-symptomatic study of some of the young family members, as is the case in individuals IV:1 (affected) and IV:2 (not affected).
DISCUSSION
Uromodulin (UMOD), or Tamm-Horsfall protein (640 amino acids, molecular weight 85-90kDa)1 expressed in the ascending loop of Henle is the most abundant protein in urine under normal conditions. Although its function has not been completely defined, it has been associated with the impermeabilisation of the distal tubule, a protective effect against urinary tract infections and renal lithiasis, as well as pro-inflammatory activity.3,4 Dysfunction in this protein has been associated with the inability to concentrate urine and tubulointerstitial fibrosis.3 Patients with a mutation of the UMOD gene have a decrease in the levels of this protein in the urine due to abnormal folding that leads to accumulation in tubular epithelial cells.5 Although the pathologies caused by mutation of the UMOD gene are not generally included in the classification of ciliopathies,6 some studies have been published in the literature that do demonstrate UMOD expression in the primary cilia.7
Mutations of the UMOD gene (chromosome region 16p2)8,9 are responsible for two tubulointerstitial nephropathies with an autosomal dominant inheritance pattern, type 2 medullary cystic disease (MCKD2) [OMIM 603860] and familial juvenile hyperuricaemia (OMIM 162000], which is currently encompassed by the term “kidney disease caused by UMOD gene mutation”.5,10-14 A gene located at locus 1q21 MCKD1 [OMIM 174000], but not identified until now,15-18 is also responsible for tubulointerstitial nephropathy with an autosomal dominant inheritance pattern. It has a similar phenotype but an earlier presentation.
Kidney disease caused by a mutation in the UMOD gene has a very low prevalence, less than 1% of end-stage kidney disease (ESKD) in adults, though it may be underdiagnosed.5 In our case series of familial nephropathies with clinical signs of hyperuricaemic interstitial nephropathy, a mutation has been detected in the UMOD gene in 12.5% of cases (unpublished data), similar to those published in the literature.19
Uromodulin deficiency causes a deficit in tubular reabsorption, favouring polyuria and hyperuricaemia.13 The fraction of excreted uric acid appears decreased early in life (even in young patients with still-normal renal function) and usually is less than 5% in adult men and less than 6% in adult women.20 Uric acid levels above the 75th percentile have been observed in more than 70% of these patients. Approximately 75% of men and 50% of women experience gout,19 but cases have also been described in which blood uric acid levels remain normal, especially in women.21 The disproportion between hyperuricaemia and the degree of renal failure is notable.19 All members of the family that were seen had hyperuricaemia since childhood and did not have a history of gout crisis.
The renal failure is slowly progressive, generally reaching the end stage between the fourth and sixth decades of life.19 The urine sediment is non-productive (proteinuria of less than 1g/day may appear in ESRD stages) and the kidney ultrasound usually reveals kidneys of reduced size and on some occasions may include medullary cysts (this has been seen in a third of patients in some series19). The levels of kidney disease and the age at presentation are highly variable, both within families and across families.13,19,22-24 In our case, all of the affected family members had hyperuricaemia and, with the exception of individual III:5, normal urine sediment. Individual III:5 had mild proteinuria of 1.1.5 mg/dl in some sediment samples. Although the majority of cases described in the literature progress without proteinuria, there are some cases of mild proteinuria.25 The high level of disease progression variability within families is notable. The age for ESRD varies from early forms (approximate age 40 years), as in the index case or in individuals I:2 and III:2, to delayed forms (approximate age 60 years), as in the case of individuals II:1 or II:4. Therefore, the diagnosis of these nephropathies may be difficult if there is a significant familial load. Familial cases that are linked to MCKD1 usually have a higher level of hyperuricaemia and more frequent and early episodes of gout, ultimately reaching end-stage renal failure earlier in life.16,26,27
On a histological level, chronic interstitial nephritis, tubular atrophy and interstitial fibrosis can be observed and often lymphocytic infiltrate. The primary lesion is thinning and progressive loss of the tubular basement membrane and the formation of cysts in the distal tubule and collector tubes. Immunohistochemical studies may reveal abnormal deposits of uromodulin in the tubular cells.11,28 In this case, the renal biopsy was only performed in 2 of the 9 cases. In the index case (individual III:4), it was performed at the time of diagnosis (20 years of age), 20 years before reaching ESKD, and non-specific changes were observed. An early renal biopsy was also performed in individual III:1 (at 29 years of age). It revealed small foci of tubular atrophy and microcystic ectasia in isolated tubules with normal electron microscopy.
The genetic study is based on sequencing the UMOD gene and detecting mutations (some 50 mutations have been described so far).3,14,19,27 Given that the majority of mutations are located on exons 3 and 4 of the gene, these are usually studied first in the index case, though mutations in other exons, such as exon 7, have also been described.19 No clear phenotype-genotype correlation has been found.13,19,29 90% of mutations are of the amino acid switch type (missense) and 62% alter the cysteine residue, leading to changes in protein folding.30 In the case of an absence of a UMOD gene mutation, analysis of the link for the MCKD1 region can be done if samples are available from other affected and non-affected family members.8,17,23
The UMOD gene is regulated by several transcription factors, among them HNF1b, whose mutation is responsible for another hyperuricaemic disease that should be suspected in cases with a compatible clinical presentation and if the studies cited above are negative.7,31,32
The primary indications for genetic diagnosis are workup for a live donor, the possibility of offering safe reproductive options and a pre-symptomatic diagnosis. The mutation found in this case has not been described previously. This is a missense-type mutation in an amino acid that is highly conserved across species with correct family segregation. The finding of this mutation allowed for the pre-symptomatic diagnosis of individual IV:1 and the disease was ruled out in individual IV:2.
There is no specific treatment for this disease nor is there for the other inherited cystic kidney diseases.5 Treatment with uricosuric medications may be helpful in reducing the progression of kidney disease,33 though the results are highly variable. Treatment with renin-angiotensin inhibitors is recommended due to the renal protective effect, though there are no clinical trials that demonstrate any specific benefits for this disease. Currently, there are no studies on specific medications for the treatment of uromodulin-associated kidney disease.
CONCLUSIONS
Kidney disease caused by mutation of the UMOD gene is not a very well known entity and, due to its high variability within and across families, it is certainly very underdiagnosed. This nephropathy should be suspected in cases of non-specific kidney disease with an autosomal dominant inheritance pattern, normal urine sediment and a kidney biopsy with predominantly interstitial fibrosis. We should also keep in mind that, as with all hereditary kidney diseases, patients with a classic phenotype tend to undergo genetic study. Therefore, the phenotypic spectrum of the disease may be much broader than has been described so far.
It is likely that advances in medical genetics may some day in the not so distant future identify the MCKD1 gene as well as other genes involved in familial interstitial nephropathy. Therefore, it is expected that understanding of the familial interstitial nephropathies will grow and result in the discovery of therapeutic targets that will someday facilitate the treatment of this entity.
Conflicts of interest
The authors declare that they have no conflicts of interest related to the contents of this article.

Table 1. Clinical characteristics of the affected families

Figure 1. Renal biopsy of the index case

Figure 2. Family pedigree

Figure 3. Sequence fragment from exon 3 of the UMOD gene



