



Hyperchloremia is a common electrolyte disorder that is associated with a diverse group of clinical conditions. The kidney plays an important role in the regulation of chloride concentration through a variety of transporters that are present along the nephron. Nevertheless, hyperchloremia can occur when water losses exceed sodium and chloride losses, when the capacity to handle excessive chloride is overwhelmed, or when the serum bicarbonate is low with a concomitant rise in chloride as occurs with a normal anion gap metabolic acidosis or respiratory alkalosis. The varied nature of the underlying causes of the hyperchloremia will, to a large extent, determine how to treat this electrolyte disturbance.
La hipercloremia es una alteración electrolítica frecuente que se asocia a una serie de distintos trastornos clínicos. El riñón desempeña una función importante en la regulación de la concentración de cloruro a través de diversos transportadores que se encuentran a lo largo de la nefrona. Sin embargo, puede aparecer hipercloremia cuando la pérdida hídrica sea mayor que la de sodio y cloruro; cuando se sobrepase la capacidad de excretar el cloruro en exceso; o cuando la concentración sérica de bicarbonato sea baja y al mismo tiempo haya un aumento de cloruro, como sucede en la acidosis metabólica con brecha aniónica normal o en la alcalosis respiratoria. La heterogénea naturaleza de las causas subyacentes de la hipercloremia determinará, en gran medida, el modo de tratar esta alteración electrolítica.
Chloride is the most abundant anion in the extracellular fluid (ECF) compartment. Hyperchloremia is defined as an increase in the chloride concentration in the plasma water. Hyperchloremia and a relative excess of chloride in the body have been linked to the development of reduced renal blood flow,1,2 increased interstitial edema including in the kidney and gastrointestinal system,3 excess morbidity and mortality in critically ill patients,4,5 and reduced survival and recovery in patients with acute kidney injury.6 Like sodium and other chemicals in the ECF compartment, chloride concentration is regulated. The organ that is responsible for the maintenance of chloride balance in the body is the kidney. This article reviews the handling of chloride by the kidney and clinical situations in which hyperchloremia can occur.
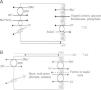
Renal handling of chlorideThe level of the chloride in the plasma is regulated by the kidney. The kidney freely filters chloride across the basement membranes of the glomeruli. The amount of chloride that is excreted into the urine is determined by the chloride filtered by the glomeruli and by a series of transport processes that occur along the nephron. Under normal circumstances, over 60% of the filtered chloride is absorbed along the proximal tubule. In the early proximal tubule, sodium is absorbed with a proportional amount of water so that the concentration of sodium does not change. By contrast, bicarbonate and other non-chloride anions are rapidly absorbed with sodium and removed from the filtrate7 (Fig. 1A). As sodium and non-chloride anions are absorbed in the early proximal tubule segments (S1 and S2), the chloride concentration in the lumen of the proximal tubule increases. By the time the tubular fluid reaches the last segment of the proximal tubule (S3), the chloride concentration is high with respect to its plasma concentration allowing chloride to be passively absorbed down its concentration gradient (Fig. 1B). The transepithelial permeability for chloride is higher than the permeability for bicarbonate so that despite the peritubular-to-lumen gradient for bicarbonate, the transport of chloride leaving the lumen exceeds the bicarbonate entering the tubular fluid.
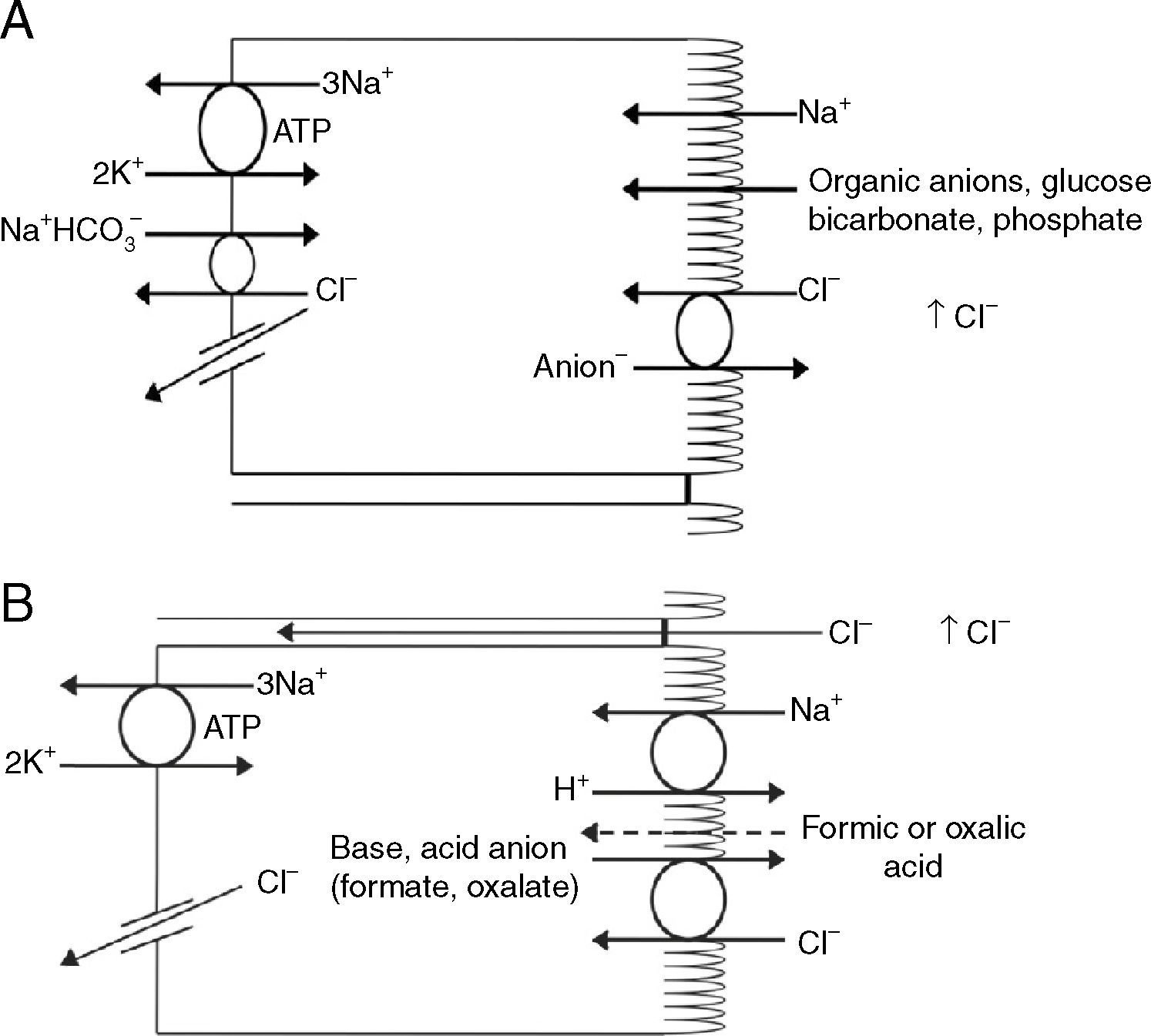
(A) In the early proximal tubule, isotonic sodium absorption occurs with organic solutes, bicarbonate, phosphate along with water resulting in a rising chloride concentration. (B) The high chloride concentration in the lumen also favors transcellular and paracellular transport. Intercellular junctions in the later proximal tubule become more permeable to chloride facilitating paracellular transport. Even when bicarbonate concentration falls in the lumen, Na+–H+ exchange continues to play a role in NaCl reabsorption. Transcellular sodium chloride absorption can occur via the coupling of Na+–H+ exchange with chloride-organic anion (formate, oxalate) exchange. The organic acid (formic or oxalic acid) is recycled into cells.
In the early portion of the proximal tubule, chloride absorption also occurs via apical chloride-anion (formate, oxalate, base) exchangers and it exits the cell via basolateral membrane transporters8 (Fig. 1B). In hyperchloremic metabolic acidosis due to HCl- or ammonium chloride-loading, the chloride reabsorption in the proximal tubule is reduced, in part, because of the reduction in organic anion transporters that facilitate sodium chloride transport9 as well as the reduction in lumen-to-peritubular gradient for chloride.
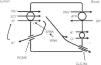
The thick ascending limb of the loop of Henle (TALH) is an important site for chloride reabsorption.10 At this site, sodium, potassium and chloride are concurrently transported via a sodium-potassium-2 chloride co-transporter (NKCC2) (Fig. 2). Chloride enters the TALH cell and leaves its basolateral aspect down an electrogenic chloride channel or via the electroneutral potassium chloride co-transporter. The movement of chloride through the basolateral chloride channel (CLC-NKB) contributes to the generation of a transepithelial positive (lumen) to negative (basolateral) potential gradient. The intracellular positive potential that would be generated by the movement of chloride out of the cell is counterbalanced by the basolateral electrogenic Na+–K+ ATPase that transports sodium out of the cell in exchange for potassium into the cell in a 3–2 ratio. ROMK potassium channels on the apical TALH cell membrane contributes to the lumen positive (intracellular negative) potential through the conductive movement of potassium ions from cell to lumen. The overall effect is that chloride, sodium and potassium enter the cell via NKCC2, and, for the most part, chloride exits the cell via the basolateral ClC-NKB chloride channel, sodium exits the cell via the Na+–K+ ATPase and potassium recycles back into the lumen via the ROMK channel or exits basolaterally via the KCl co-transporter. The tight coupling between sodium and chloride transport in the TALH is underscored by one of the varieties of Bartter syndrome in which defects in basolateral chloride channels disrupt sodium chloride reabsorption and mimics the renal defect observed with abnormal NKCC2 proteins. Although other transporters on the peritubular side of the TAL cell such as the KCl co-transporter will transport chloride in a sodium-independent manner, most of the chloride that is absorbed by the TALH is coupled with sodium reabsorption. Therefore, factors that increase sodium reabsorption in this segment will also increase chloride reabsorption.
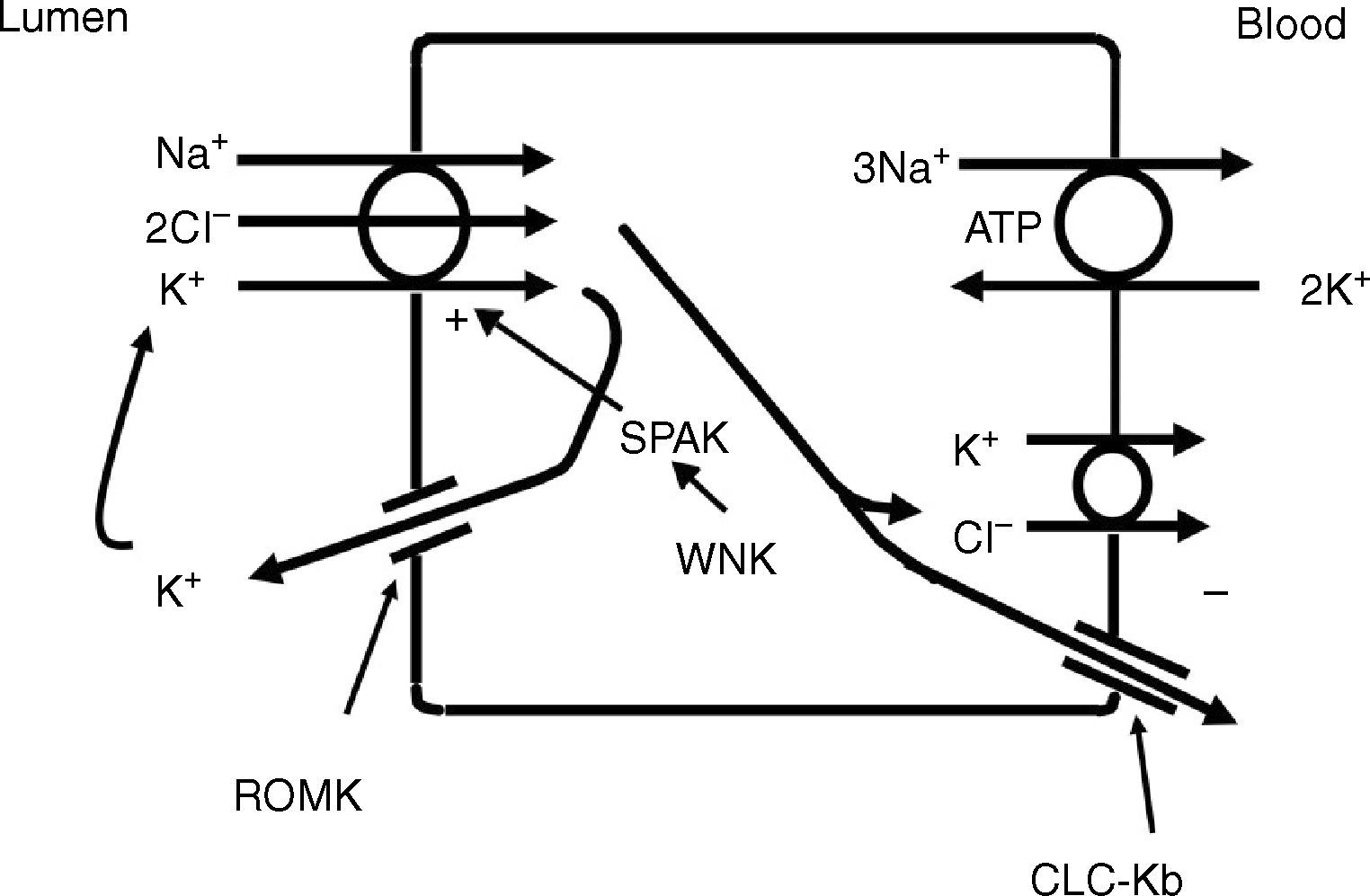
The thick ascending limb of the loop of Henle absorbs chloride via the apical Na+–K+–2Cl− cotransporter (NKCC2) and chloride exit from the cell via a basolateral chloride channel and by K+–Cl− cotransport. K+ recycling into the lumen and basolateral conductive Cl− exit via CLC-Kb contribute to the positive-to-negative lumen to basolateral transepithelial gradient. Intracellular chloride may regulate NKCC2 transport through a chloride-sensing WNK kinase (WNK) that can activate the STE20/SPS1-related proline/alanine rich kinase (SPAK) and NKCC2 when intracellular Cl− is low. On the other hand, when chloride accumulates in the cell due to defects in basolateral chloride channel exit pathway, NKCC2 transport is blocked. When NKCC2 is stimulated, for example by antidiuretic hormone, chloride entry is increased, but basolateral Cl-conductance is also enhanced.
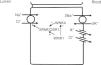
In the distal convoluted tubule, sodium and chloride are transported from the lumen into the cell by a sodium-chloride co-transporter (NCC)11 (Fig. 3). The driving force for movement of chloride from lumen into cells comes from the lumen-to-cell sodium gradient that is generated by the basolateral Na+–K+ ATPase which pumps sodium out of the cell thereby maintaining low intracellular sodium concentrations. Further regulation of NCC and NKCC may occur through WNK kinases, which may serve as chloride sensors12 and can regulate these transporters by modifying trafficking or their phosphorylation state.13 In later portions of the distal convoluted tubule, a negative lumen potential generated by movement of sodium through the apical epithelial sodium channel (ENaC) can also serve as a driving force for passive chloride reabsorption. Thus, the segments of distal convoluted tubule display direct coupling of sodium and chloride transport via the NCC and indirect coupling of transport via passive movement down an electrochemical gradient.
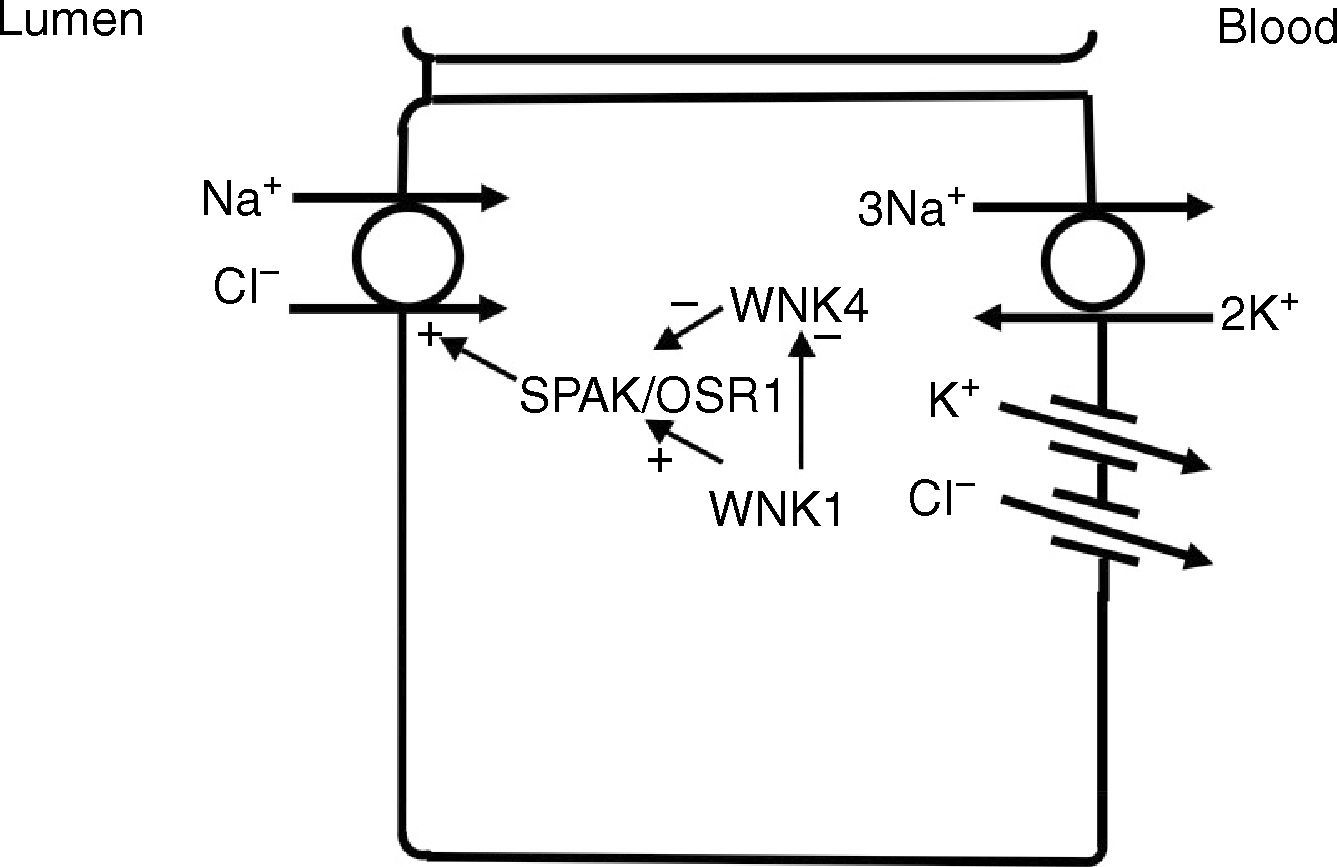
In the distal convoluted tubule, sodium and chloride in the lumen are taken up into the cell via a Na+–Cl− cotransporter (NCC). Transport via NCC is driven by a low intracellular sodium mostly generated by the basolateral Na+–K+ ATPase. The WNK1 kinase may serve as a chloride sensor to block inhibition of NCC by the WNK4 kinase.
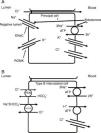
The collecting duct plays an important role in determining the chloride content of the final urine. Chloride reabsorption in this portion of the nephron helps to conserve chloride in response to low chloride intake and can contribute to the hypertensive effects of a high sodium chloride diet. Most of sodium that is reabsorbed in the collecting duct occurs in principal cells via aldosterone-regulated apical epithelial sodium channels. Chloride reabsorption in the collecting duct can occur via paracellular chloride absorption that is driven by the lumen negative transepithelial potential generated by lumen-to-cell sodium flow through ENaC (Fig. 4A). In addition, in B-type and non-A non-B type intercalated cells, chloride can be transported via pendrin, a chloride-bicarbonate exchanger, with chloride moving from lumen-to-cell while bicarbonate secreted into the lumen (Fig. 4B). The relationship between various sodium and chloride transport processes in this portion of the nephron was illustrated in a recent paper by Vallet and colleagues.14 The authors performed a number of physiological maneuvers to determine their effects on ENaC and pendrin protein levels in the kidney. Long-term NaCl loading significantly decreased pendrin protein levels while there was a decrease in the “active” ENaC-γ submit and an enhanced β-subunit. A dissociation between sodium and chloride transport was observed, however, with the inhibition of the sodium-chloride co-transporter with hydrochlorothiazide, pendrin levels fell but ENaC levels increased. NaCl restriction increased pendrin expression.15 An enhanced luminal bicarbonate concentration that would be created by pendrin-mediated bicarbonate secretion affects sodium reabsorption by increasing the activity of downstream ENaC.16 Sodium chloride transport by intercalated cells can also be enhanced by the presence of a thiazide-sensitive apical sodium-dependent chloride-bicarbonate exchanger (NDCBE, Slc4A8) which transports 1 sodium and 2 bicarbonate ions from the lumen into the cell in exchange for 1 chloride ion which leaves the cell. If NDCBE transport is coupled with pendrin-mediated chloride-bicarbonate exchange, the two transporters working together could result in net sodium chloride reabsorption from the lumen, as the bicarbonate recycles into and out of the cell while sodium and chloride enter the cell17 (Fig. 4B). Factors which alter the ratio of the amounts or activities of these two anion exchangers may determine the net impact on bicarbonate secretion and chloride reabsorption. Another transporter which may be involved in excretion of excessive chloride in the body is the Slc26A9 transporter which may act as a chloride channel in the medullary portions of the collecting duct.18 It may modify the impact of chloride loads by increasing chloride secretion under conditions of chloride excess. Knockout of this gene results in a predisposition to hypertension. Mice deficient in this protein develop hypertension when exposed to a high sodium chloride load.18 Although the Slc26a9 transporter appears to play an important role in handling large sodium chloride loads, the regulation of the native transporter activity in response to varied sodium chloride loads remains unknown.
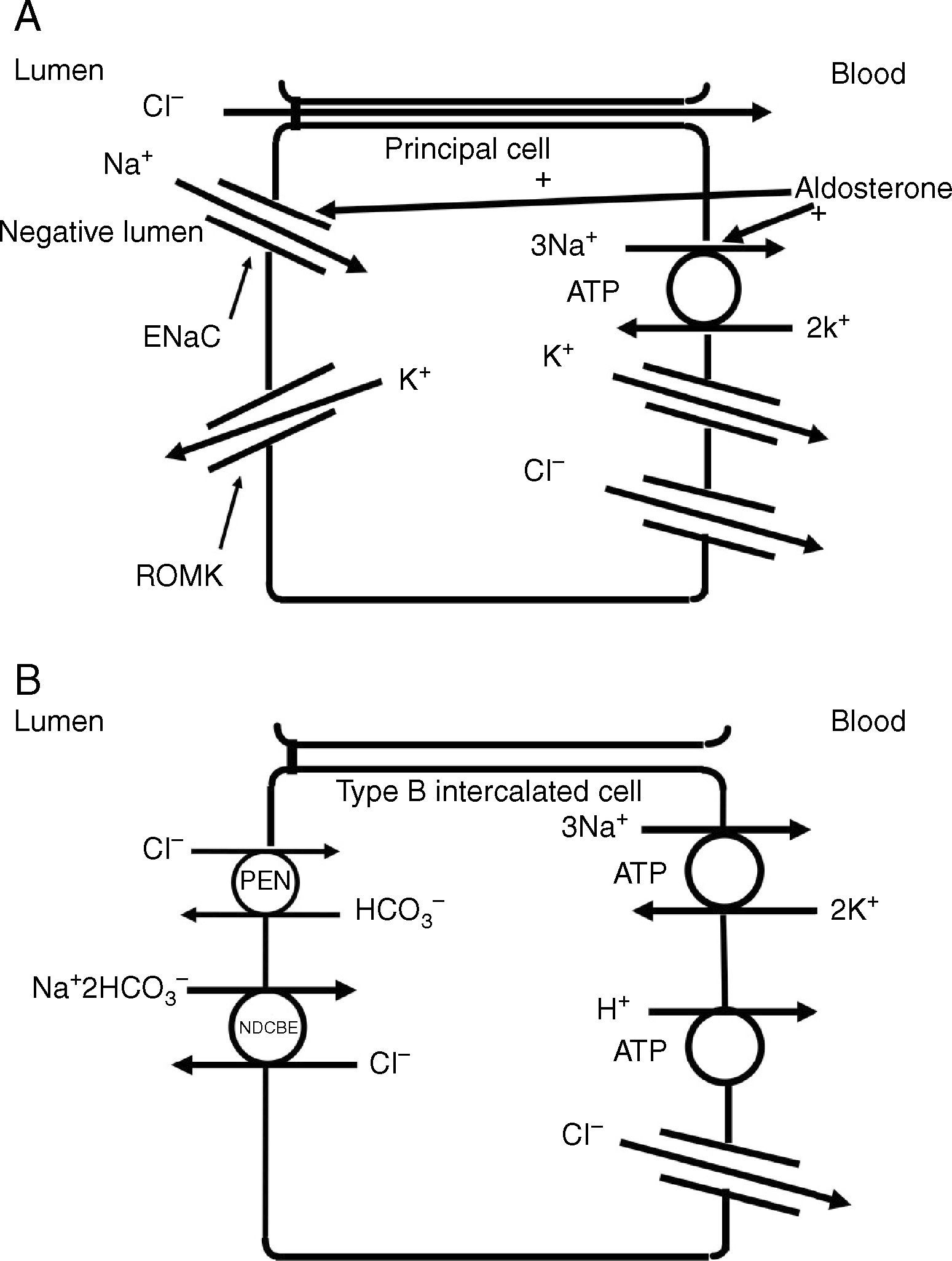
(A) Chloride can be secreted or reabsorbed in the collecting duct. A portion of chloride absorption is driven by a lumen negative potential and paracellular movement. (B) Transcellular reabsorption of chloride can also occur via coupling of the apical Pendrin chloride-bicarbonate exchanger to the SLCA48 sodium -dependent chloride-2 bicarbonate exchanger (NDCBE). 2 cycles of Pendrin would result in 2 chlorides entering the cell in exchange for 2 bicarbonate while NDCBE would transport 1 sodium and 2 bicarbonates in exchange for 1 chloride out. The net result would be the transport of 1 sodium and 1 chloride into the cell. Differences in the apical Pendrin and NDCBE activities could determine whether Cl− secretion or absorption predominates.
The serum chloride level is generally measured as a concentration of chloride in a volume of serum. The biologically active chloride concentration is the concentration of free chloride in the plasma water. Chloride is most frequently measured by using a silver-chloride electrode either in a direct or diluted serum sample.19 Automated methods found in many laboratories involve the dilution of the serum sample with reagent so that the volume of the sample is assumed to have a normal water content and estimations are made based upon assuming a normal dilution factor. When the solid components of the serum are very high, as can occur with hypertriglyceridemia and multiple myeloma, pseudohypochloremia can occur. Pseudohyperchloremia can also be seen in bromide or iodide intoxication. The interaction of bromide or iodide with the silver-chloride electrode generates a greater voltage change than does chloride giving the impression of excessive chloride in the blood.20,21
Causes of true hyperchloremiaHyperchloremia from water lossHyperchloremia can result from a number of mechanisms (Table 1). Water loss in excess of chloride loss can raise the chloride concentration.22 In dehydration, the renal response is to conserve water and lower urine output. As there may also be a component of volume depletion with more severe degrees of dehydration, conservation of chloride as well as sodium occurs via increased proximal tubule reabsorption of chloride and other solutes, and reduced delivery of chloride and sodium to more distal nephron segments. The enhanced proximal tubular reabsorption of tubular fluid and its contents will not necessarily change the chloride concentration as the absorption of fluid occurs isotonically. The treatment of water deprivation is the judicious administration of electrolyte-free water which will reduce both the sodium and chloride concentrations.
Causes of hyperchloremia.
| Pseudohyperchloremia |
| High amounts of serum solids (lipids or proteins) when assays involving sample dilution are used |
| Bromide or iodide intoxication |
| Excessive chloride administration |
| Large volume administration of 0.9% (normal) sodium chloride solution |
| Administration of hypertonic saline |
| Saltwater drowning |
| Net water losses |
| Fever |
| Perspiration |
| Inadequate water intake (poor thirst or access to water) |
| Diabetes insipidus |
| Loss of water in excessive electrolytes |
| Certain forms of diarrhea |
| Osmotic diuresis |
| Certain cases of post-obstructive diuresis |
| Associated with metabolic acidosis |
| Certain forms of diarrhea |
| Renal tubular acidosis |
| Carbonic anhydrase inhibitors |
| Ureteral diversion (e.g., ileal bladder) |
| Ammonium chloride administration |
| Arginine or lysine hydrochloride administration |
| Certain cases of chronic kidney disease |
| Organic acidosis in which acid anion is rapidly excreted (e.g., toluene overdose) |
| Respiratory alkalosis |
Hyperchloremia can occur when the body is exposed to fluids that are high in chloride. An extreme example of this is salt water drowning/ingestion. The sudden large input of seawater (average salinity is 3.5%) overwhelms the ability of the kidney to excrete the sodium chloride load and hypernatremia and hyperchloremia are common.23 Nevertheless, a component of the hypernatremia and hyperchloremia associated with excessive saltwater ingestion comes from fluid losses associated with diarrhea and urinary losses.23 The treatment of patients with hyperchloremia from saltwater drowning will depend upon the patient's volume status as well as estimates of ongoing fluid and electrolyte losses and the judicious replace of water and electrolytes as needed.
A less extreme example of hyperchloremia with an excessive sodium chloride load is the administration of large volumes of isotonic (0.9%) sodium chloride solution (normal saline) frequently used for volume resuscitation of patients. It is noteworthy that when a normal individual is given a large bolus of isotonic saline, it may take up to 2 days to return to the pre-treatment state of sodium and chloride balance.24 This retention of chloride occurs with exposure to the supraphysiologic levels of chloride in normal saline. The normal chloride concentration in the plasma is in the 95–110meq/L range, while normal saline has a chloride concentration of 154meq/L. The relatively slow excretory response to isotonic saline may be related to effects of chloride loads on renal blood flow and on glomerular filtration (tubuloglomerular feedback). Although down-regulation of chloride reabsorptive transporter activities occur with sodium chloride loading,14,25,26 the rapidity of the reduction of these transporters is not well defined.
With isotonic saline administration, the bicarbonate concentration may also fall as the chloride concentration rises. Besides dilution of the plasma bicarbonate with administration of supraphysiologic chloride-containing, base-free solutions such as normal saline, other factors may play roles in the fall in bicarbonate and rise in chloride levels. Urinary bicarbonate losses may contribute to the fall in serum bicarbonate level as there may be a reduction in the reabsorptive threshold for bicarbonate with volume expansion.27 This loss of bicarbonate can occur even when the serum bicarbonate concentration is low.27 In studies in humans, over the first 24h after an infusion of isotonic saline is given, the losses of sodium and potassium exceed those of chloride. The reduced excretion of chloride in comparison to sodium and potassium suggested the urinary loss of other anions such as bicarbonate and other organic anions that may also contribute to a fall in the serum bicarbonate concentration.24 Using balanced electrolyte solutions that contain base or base-equivalents and chloride concentrations that are more physiologic may not only prevent the development of hyperchloremic acidosis, but may avoid some of the possible harmful effects of associated with hyperchloremic solutions such as normal saline.28,29 In comparison with balanced, base-containing salt solutions, normal saline administration to healthy human subjects resulted in a fall in renal blood flow and cortical perfusion30 raising concerns about the excessive administration of normal saline in volume resuscitation in patients. Nevertheless, certain clinical situations may favor the use of normal saline including in patients with hypochloremic metabolic alkalosis or those with cerebral edema.
Hyperchloremia with metabolic acidosisHyperchloremia also occurs when hydrochloric acid (HCl) is added to the blood. HCl is rarely given as a direct acidifying agent but can be created from the metabolism of ammonium chloride or cationic amino acids such as lysine and arginine.31 The generation of HCl leads to reaction of H+ with HCO3− that results in CO2 production and a net loss of HCO3− and rise in chloride concentration.
With respiration titrated bicarbonate is lost from the body as CO2.Thus for every milliequivalent of HCl added, a milliequivalent of bicarbonate is consumed and converted to CO2 so that the chloride level rises to the same extent as the bicarbonate level falls.
Renal tubular acidoses (proximal type 2 RTA and distal type 1 or 4 RTA) result in hyperchloremic metabolic acidosis. In proximal RTA (type 2), bicarbonate reabsorption in the proximal tubule is impaired resulting in increased losses of bicarbonate out of this segment. There is also some disruption of chloride reabsorption because the lack of the extraction of bicarbonate prevents the normal rise in luminal chloride concentration. Nevertheless, in proximal RTA, the reduction in bicarbonate transport is greater than the reduction in chloride transport so that there is relatively more chloride reabsorbed than bicarbonate. If carbonic anhydrase inhibition is used as a model for proximal RTA, chloride reabsorption appears to be less impaired than bicarbonate reabsorption as is reflected by a relatively modest increase in the urinary chloride excretion rate while the rates of excretion of sodium, potassium and, presumably, bicarbonate are markedly increased.32
In classical distal RTA (type 1) or type 4 RTA, the reduction in net acid secretion prevents the renal generation of new bicarbonate by impairing ammonium and/or titratable acid excretion. As a result, the HCl generated by metabolism results in a fall in bicarbonate that is not compensated for by the generation and conservation of bicarbonate and excretion of chloride. As long as renal function is preserved, non-chloride acid anions do not accumulate in the systemic circulation maintaining a relatively normal anion gap. Indeed, the renal excretion of phosphate and sulfate anions generated from the metabolism of phosphorus- and sulfur-containing amino acids31 is actually stimulated by acidosis.33
Another cause of hyperchloremic metabolic acidosis occurs with diarrhea. In many segments of the gastrointestinal tract and associated exocrine organs such as the pancreas, bicarbonate is secreted into the gut in exchange for chloride so that loss of bicarbonate, especially in secretory forms of diarrhea, can be associated with bicarbonate losses which are associated with chloride retention.34
Repair of hyperchloremic forms of metabolic acidosis involves stopping the ongoing cause of bicarbonate loss or HCl generation while giving the patient bicarbonate or base equivalents (e.g., citrate) or allowing the patient's kidneys to regenerate bicarbonate if renal function is relatively normal. During the generation of metabolic acidosis, there are initially net sodium losses and volume contraction. With more prolonged acidosis, there may be sodium retention due to high aldosterone levels and upregulation of ENaC in the collecting duct.35 With provision of bicarbonate to correct the acidosis, bicarbonate is retained in the proximal tubule and normal chloride reabsorption is also re-established. The associated volume re-expansion with bicarbonate may contribute to the fall in chloride. When the kidneys repair the metabolic acidosis, ammonium chloride is excreted in the urine while bicarbonate that is made in the proximal tubule as a byproduct of the glutamine metabolism is returned to the blood.
List of key points- 1.
The kidney plays a key role in maintaining chloride balance in the body. Although renal chloride transport is coupled with sodium transport, chloride transport may sometimes diverge from sodium transport.
- 2.
Hyperchloremia can result from a variety of conditions including water depletion, excessive chloride exposure and metabolic acidosis.
- 3.
The pathogenic cause of hyperchloremia will provide guidance on how the disturbance should be treated: water depletion is treated with judicious water repletion; excess chloride administration by withholding further chloride administration; and hyperchloremic metabolic acidosis by giving bicarbonate.
The author declares no conflict of interest.
This work was supported, in part, by the U.S. Department of Veterans Affairs.



