






Secondary hyperparathyroidism (SHPT) is an integral component of the chronic kidney disease-mineral and bone disorder (CKD-MBD). Many factors have been associated with the development and progression of SHPT but the presence of skeletal or calcemic resistance to the action of PTH in CKD has often gone unnoticed. The term hyporesponsiveness to PTH is currently preferred and, in this chapter, we will not only review the scientific timeline but also some of the molecular mechanisms behind. Moreover, the presence of resistance to the biological action of PTH is not unique in CKD since resistance to other hormones has also been described (“uremia as a receptor disease”). This hyporesponsiveness carries out important clinical implications since it explains, at least partially, not only the progressive nature of the pathogenesis of CKD-related PTH hypersecretion and parathyroid hyperplasia but also the increasing prevalence of adynamic bone disease in the CKD population. Therefore, we underline the importance of PTH control in all CKD stages, but not aiming to completely normalize PTH levels since a certain degree of SHPT may represent an adaptive clinical response. Future studies at the molecular level, i.e. on uremia or the recent description of the calcium-sensing receptor as a phosphate sensor, may become of great value beyond their significance to explain just the hyporesponsiveness to PTH in CKD.
El hiperparatiroidismo secundario (HPS) es uno de los componentes integrales de las alteraciones del metabolismo óseo-mineral en la enfermedad renal crónica (ERC) o complejo CKD-MBD («Chronic Kidney Disease-Mineral Bone Disorder»). Se ha demostrado que en el desarrollo y progresión del HPS intervienen muchos factores, estrechamente interrelacionados, pero la presencia e importancia de hiporrespuesta (o resistencia) a la acción de la hormona paratiroidea (PTH) es poco comprendida. En esta revisión analizaremos sus antecedentes, factores que intervienen, así como alguno de los mecanismos moleculares que podrían explicarla. La presencia de resistencia a la acción biológica de la PTH no es única en la ERC ya que también se presenta para otras hormonas, habiéndose incluso usado el término de “uremia como una enfermedad de receptores”. Esta hiporrespuesta a la PTH tiene importantes implicaciones clínicas, dado que no sólo permite explicar parte de la patogenia progresiva de la hipersecreción de PTH e hiperplasia paratiroidea, sino también la creciente prevalencia de enfermedad ósea adinámica en la población con ERC. De este modo, subrayamos la importancia de controlar, sin normalizar completamente, los niveles de PTH en los distintos estadios de ERC dado que un cierto incremento de sus niveles supone inicialmente una adaptación clínica. Futuros estudios a nivel molecular sobre la uremia o la reciente descripción del efecto directo del fosfato sobre la actividad del receptor sensor de calcio como sensor de fosfato, podrían resultar valiosos incluso más allá de explicar la hiporrespuesta a la PTH en la ERC.
Chronic kidney disease (CKD) is a major global health problem with an extremely high risk of cardiovascular disease and a large increase in mortality.1,2 This is partly related to disturbances in bone mineral metabolism (chronic kidney disease-mineral bone disorders [CKD-MBD]). The CKD-MBD syndrome/complex includes biochemical abnormalities. bone disorders and/or cardiovascular calcifications; all of them closely interrelated.3 On the other hand, the term “renal osteodystrophy” is currently considered only a measure of the bone disorder measurable by histomorphometry.3 Currently, we have to consider bone as an endocrine organ since it produces hormones with systemic actions such as fibroblast growth factor 23 [FGF23], sclerostin and osteocalcin, all potentially responsible for metabolic and cardiovascular complications observed in CKD patients.4
The progressive increase in parathyroid hormone (PTH) levels (secondary hyperparathyroidism [SHPT]) is one of the components of the CKD-MBD complex and, if left untreated, will worsen biochemical disturbances (i.e. hypercalcemia and/or hyperphosphatemia), bone structure (high turnover bone disease) and will be associated with cardiovascular disorders, among other effects.5,6 Classically, PTH has been considered an “uremic toxin” with effects not limited to bone.7,8 In fact, PTH induces an increase in intracellular calcium (Ca) in different cell types and stimulates the production of FGF23, which has also systemic effects. Improvement of some of these adverse effects after parathyroidectomy (PTX) supports a causal link, direct or indirect, with the elevated PTH levels.8,9 These potential toxic effects of PTH could also explain the association of SHPT with the progression of CKD, atheromatous and non-atheromatous cardiovascular disease and mortality, as well as its association with all-cause mortality.10–12 Of note, beyond CKD, some cohorts of cardiological patients have confirmed the independent association between high PTH levels, cardiovascular events and mortality (including sudden death).13
SHPT is a classic and common complication of CKD that is progressive, and potentially serious. However, there is also evidence that very low or relatively low levels of PTH have also been associated with high morbidity and mortality.10,14,15 Low PTH is responsible of low bone remodeling (its most frequent manifestation is currently adynamic bone disease [ABD]).16 This low bone remodeling has been recently associated with morbidity and mortality, attributed to an increase in bone fractures or microfractures, and/or the appearance or progression of cardiovascular calcifications.15,17–23 Therefore, it is not surprising the complexity of the known association between mortality and PTH, only evident at both extremes (high and low PTH levels).3,10 Cohort studies show that only extreme PTH levels can predict, with acceptable sensitivity/specificity, the underlying bone turnover [low bone turnover (ABD) or high bone turnover (osteitis fibrosa)].24 The combination of PTH with alkaline phosphatase levels (especially its bone fraction) could improve this predictive capacity, but it is still far from the “gold standard” which is bone biopsy.25–28
Pathophysiology of secondary hyperparathyroidismThe progressive loss of renal function produces a decrease in the expression of the PTH receptor (PTHR1) and α-klotho in the kidney, and also a reduction of the PTHR1 in the bone. There is also an elevation of FGF23 that helps to increase urinary excretion of P, but the high FGF23 levels cannot avoid retention of P if renal function is markedly reduced.29–31 The increased FGF23 reduces the synthesis and increases the catabolism of calcitriol (1,25-dihydroxyvitamin D; active vitamin D), among many other metabolic disorders.32,33 All these factors, extensively reviewed in other articles,32,34,35 involve several interrelated mechanisms, including the decreased expression of receptors in parathyroid cells: the Ca sensing receptor (CaSR), the vitamin D receptor (VDR), the FGFR1 receptor and klotho; all these receptors are responsible for the inhibition of synthesis and secretion of PTH as well as its capacity to proliferate. With the reduction of the expression of these receptors in the parathyroid glands hyperplasia develops; it is initially polyclonal in nature, and it potentially becomes monoclonal if pro-proliferative stimuli persist. It is known that parathyroid hyperplasia develops progressively from the initial stages of CKD7,32,36 and in advanced CKD, despite the increase in FGF23 and PTH, renal P excretion is not sufficient to maintain normal P balance. Consequently, there is an increase in extracellular P which becomes both a direct and indirect stimulus for PTH secretion. Hyperphosphatemia prevents the activation of the CaSR which stimulates the secretion of PTH.37–40 In addition high P is partly responsible for a reduced calcemic response to PTH, favoring hypocalcemia, the main stimulus for parathyroid gland (PTG) proliferation.
Extracellular Ca is the most important regulator of parathyroid gland function.8,36 To correct hypocalcemia, the stored PTH is rapidly secreted, and this is followed by an increase in the synthesis of new PTH. Finally, if the hypocalcemic stimulus remains, an increase in the number of functioning parathyroid cells is required and consequently hyperplasia develops. In fact, both the previously mentioned hyperphosphatemia and reduction of calcitriol will secondarily cause hypocalcemia through two different mechanisms, reduced Ca efflux from bone (hyperphosphatemia), and a decrease in the intestinal Ca absorption (calcitriol deficiency), thus contributing to the development of SHPT. In the parathyroid cell, sustained hypocalcemia reduces the expression of receptors that inhibit its activity (CaSR, VDR, FGFR1-klotho), increasing the synthesis and secretion of PTH, and inducing parathyroid cell proliferation.34,35,41,42 In this review, beyond analyzing the complex pathophysiological interactions among the different factors leading to SHPT, we will focus on the importance of hyporesponsiveness (calcemic or skeletal resistance) to the action of PTH in the pathogenesis of SHPT and to what extent it can contribute to the development of ABD.
Resistance or hyporesponsiveness to the action of parathyroid hormoneResistance to the action of PTH in CKD, (also known as skeletal resistance, bone resistance, decreased calcemic response, or simply resistance to PTH) is an old concept,43 currently renamed as “hyporesponsiveness” to PTH.44 In fact, both bone and kidney responses to the action of PTH are progressively impaired in CKD44 and the term “hyporesponsiveness” may be more appropriate, since the response to PTH is mitigated but it is not completely absent44 (Table 1).
Terminology.
| Hyporesponsiveness to PTH | Current generic term expressing a decrease in the normal biological response to the action of the own (endogenous) PTH that occurs in CKD. It may be brought out as a decrease of the calcemic response to a PTH infusion (in experimental models) or as a bone with a decreased remodeling relative to the amount of PTH (for instance in bone biopsies). Hyporesponsiveness implies a decreased number or a dysfunction of bone cells. |
| Decreased calcemic (skeletal) response to the action of PTH or resistance to the PTH | Experimental demonstration of the presence of hyporesponsiveness to PTH. The infusion of a fixed amount of PTH into the experimental animal induces an expected increase in plasma calcium. The degree of increase in calcium in response to PTH will be reduced by the presence of different factors (degree of kidney function, phosphorus content in the diet, etc.). Experimental animals receive a diet without calcium during the PTH infusion so that the increase in plasma calcium is considered a measure of the action of PTH on the skeleton. |
CKD: chronic kidney disease; PTH: parathyroid hormone.
Hyporesponsiveness to PTH was first described by JM Evanson in 196643 in 12 hypocalcemic patients with CKD.43 Thereafter, Massry et al.45 observed that the calcemic response to a parathyroid extract was markedly decreased in parathyroidectomized dogs after the induction of uremia, and that the calcemic response to PTH was reduced in patients with moderate and advanced CKD (including patients on hemodialysis and kidney transplantation).46 Llach et al. described a decreased calcemic response to endogenous PTH.47,48 These observations indicated that the decreased calcemic response to PTH occurs at early stages of CKD; consequently, a higher concentration of circulating PTH is required to maintain Ca homeostasis. Since the old study by Albright et al.49 and the “trade-off hypothesis” by Bricker and Slatopolsky,30,50 it had been proposed that the retention of P and the reciprocal decrease of Ca would induce parathyroid hyperplasia and osteitis fibrosa. The presence of skeletal resistance to PTH in CKD would provide an additional, generally forgotten, amplifying mechanism in the generation of hypocalcemia and the progressive development of SHPT in CKD.32,51,52 The presence of skeletal resistance to the action of PTH at the initial stages of CKD would contribute, along with other factors (hyperphosphatemia, vitamin D deficiency, etc.), to the development of hypocalcemia, which in turn would stimulate the parathyroid glands, increasing PTH secretion and/or inducing glandular hyperplasia. The elevated PTH level would maintain a normal serum calcium concentration during early CKD. On the other hand, the late onset of hypocalcemia, only detectable in advanced stages of CKD, would clearly represent an end stage demonstrating the inability of PTH to restore Ca to normal levels, thus reflecting the maximum clinical expression of hyporesponsiveness to PTH.
Factors linked to the hyporesponsiveness to parathyroid hormone in chronic kidney diseaseThere are multiple factors that have been associated through different mechanisms to the hyporesponsiveness to PTH in CKD. These are factors related to alterations in mineral metabolism in the context of CKD or the undefined effects of the accumulation of (known or unknown) uremic toxins in more advanced stages of CKD (“uremia”). Various demographic features (race, age and sex) or pathological conditions such as diabetes, among many others, may play a role (Fig. 1).
Decreased calcitriol levels
The very first work in this field considered that vitamin D was necessary for an adequate effect of PTH on bone43; subsequently, it was demonstrated that administration of calcitriol restored, at least in part, the calcemic response to PTH in experimental animals with renal failure.53,54 This observation led to the conclusion that the reduced calcemic response to PTH would be associated with decreased levels of calcitriol during the initial stages of CKD. Furthermore, in patients with early CKD, the daily administration of calcitriol for 6 weeks improved the calcemic response to PTH.48 Finally, experimental studies also showed an improvement after the administration of calcitriol together with 24.25 (OH)2D3.55 Therefore, although the underlying mechanism was not clear, it would appear that vitamin D could enhance the action of PTH on bone. However, other researchers did not confirm this effect.53,56
Phosphate retentionFrom the classic works by Somerville and Kaye, as well as others performed in different experimental models,54,57 it is well known that P retention significantly decreases the calcemic response to PTH in CKD. Since high P inhibits calcitriol synthesis in part through an elevation in FGF23, it cannot be excluded that some of the resistance to the calcemic action of PTH attributed to high P could be indirectly mediated by the reduction in calcitriol production. It was also shown that rats with CKD, fed a low-P diet, had an improved calcemic response to a constant standardized infusion of PTH 1–34, but only rats with moderate CKD had a significant concomitant increase in calcitriol levels. Thus, P restriction improved the calcemic response to PTH but, in advanced CKD, this beneficial effect was independent of calcitriol. In fact, in subsequent studies we demonstrated that the negative effect of P retention on the calcemic response to PTH may be much greater than the effect of calcitriol deficiency.58,59 The improvement of the calcemic response to a PTH infusion after P restriction has also been demonstrated in patients with mild CKD.60
Interestingly, in rats with normal kidney function which received a high P diet, the calcemic response to PTH was reduced in the absence of changes in serum P concentration, indicating that the content of P in the diet itself was responsible, directly or indirectly, for the reduction of the calcemic response.54,61 This is an issue to be currently taken into account, given the lack of a clear recommendation on whether the restriction of P in the diet should be applied in CKD stages G2-G3. The intrinsic mechanism that leads to a decrease in the calcemic response to PTH induced by P is not fully understood, but it is likely that, in addition to the negative effect of P on calcitriol levels, the environmental P concentration in bone could affect the amount of exchangeable Ca from bone that could be mobilized by PTH.53,62
All of the aforementioned experiments were performed prior to the discovery of FGF23, Dickkopf-1 (Dkk1) or sclerostin. To date, the possibility that high levels of these molecules could reduce the effect of PTH on bone has not been excluded. It could be that FGF23, directly or indirectly through the suppression of calcitriol, or the activation of Dkk1 or sclerostin (inhibitors of the Wnt/ß-catenin pathway) could interfere with the flow of Ca from bone mediated by PTH.44 Recent reports have also shown that the CaSR possess specific binding sites for P that interfere with CaSR activation by Ca2+63,64 (P sensor). Therefore, while Ca2+ activates the CaSR, PO43– partially prevents its activation contributing to a greater secretion of PTH.63 In the same way that P interferes with the activation of CaSR in the parathyroid cell, it could also interfere with the CaSR of osteoblasts and osteoclasts, but this has not been investigated. In short, the interaction of P on bone CaSR could interfere with the calcemic response to PTH in CKD.
Although both P restriction and calcitriol administration improve the impaired calcemic response to PTH, none fully restored it, neither alone nor in combination. In contrast, in PTX or thyroiparathyroidectomized animals,54,56,65 the elimination of circulating PTH surprisingly corrects the calcemic response to PTH, even despite the presence of hyperphosphatemia and low calcitriol levels.
Downregulation of parathyroid hormone receptorsIt has been previously described that in uremic animals the elevation of endogenous PTH could desensitize the skeleton to the administration of exogenous PTH through downregulation of bone receptors (as a defense mechanism).56,66 The potential role of downregulation of PTH bone receptors was postulated after the “surprising” restoration of the calcemic response to PTH after PTX. Likewise, in bones from uremic dogs, with acute or chronic renal failure, there was a reduction in cyclic AMP release in response to PTH administration, and it was corrected after thyroparathyroidectomy.67,68
Although in experimental animals PTX restores the calcemic response to PTH, we observed that maintaining PTH levels in the normal range in CKD rats fed a low P diet did not restore the calcemic response.65 Furthermore, uremic animals with normal levels of PTH, obtained after partial PTX, still showed a 50% decrease in the calcemic response to PTH compared to normal rats.65 This is consistent with clinical studies that had shown that subtotal PTX nearly normalized PTH levels, but did not improve the calcemic response to PTH.46 Additionally, PTX improved the calcemic response to PTH not only in animals with CKD, but also in control animals.65 Therefore, down-regulation of PTH receptors after excessive exposure does not seem to be the only explanation for the restoration of the calcemic response after PTX. In addition to the classical explanation that the restoration of the calcemic response to PTH after PTX is due to a phenomenon of hypersensitization, also described in other hormonal systems, it is possible that PTX could also affect the accumulation or distribution of available Ca in bone, as described in other studies.69
All these findings do not exclude downregulation of PTH receptors as a potential cause of decreased calcemic response to PTH.70 The cloning of the PTH receptor type 1 gene (PTHR1), a common receptor for PTH and PTH-related peptide (PTHrP),71,72 has allowed to show that PTH1R is widely distributed in tissues,73 and that it is downregulated in uremic kidneys and bone.74–77 Human data is less consistent since studies by different authors have shown both a decrease and an increase in bone expression of PTH1R. This apparent discrepancy may be explained by the use of different methodologies and dissimilar characteristics of the populations studied.
On the other hand, the finding of down-regulation of PTH1R messenger RNA (mRNA) in bone tissue and not in the liver or heart suggests that PTHR1 expression is regulated in a specific manner by each cell, regardless of the uremic state.78 It is likely that other factors, in addition to increased PTH levels, could be responsible for the decrease in receptor activity.75,78 Thus, it has been shown that neither the increase in PTH and P nor the decrease in plasma Ca play an important role in the down-regulation of renal PTH1R in CKD, and it would also seem unlikely that it is secondary to an increase in renal PTHrP.75 However, other authors observed a higher expression of PTH1R mRNA after PTX, but no controls were included.76
Although the mechanisms responsible for the presumed desensitization or downregulation of PTH1R in CKD remain poorly defined, some studies have implicated several uremic factors and C-terminal PTH fragments (see below) in this phenomenon. The available information on the regulation of PTH1R is very limited and even contradictory, and it is certainly beyond the scope of this review.79
Uremic toxinsIt is important to highlight that, in rats with CKD, we observed a significant decrease in the calcemic response to a PTH infusion despite the presence of normal serum levels of Ca, P, PTH and even calcitriol (Fig. 2). This calcemic response directly reflects the skeletal hyporesponsiveness to the action of PTH, since the animals receive a calcium free diet during the infusion. This finding allowed us to infer that factors intrinsic to uremia per se (identified or non-identified uremic toxins) decreased the calcemic response to PTH.65 These findings were confirmed using a different model of PTX uremic rats with normal PTH that received a constant infusion of PTH and the calcemic response was neither corrected.80
![Hyporesponsiveness to the action of parathyroid hormone (PTH). The calcemic response (plasma calcium [Ca] in mg/dl) to the infusion for 48 h of a constant amount of PTH in rats with different degrees of renal function (normal, moderate renal failure and advanced renal failure) and different content is shown of phosphorus in the previous diet (PA: high phosphorus diet [1.2%]; BP: low phosphorus diet [0.3%]; MP: moderate phosphorus diet [0.6%]) rats do not receive calcium in the diet, so the increase in calcium is due to the skeletal response to the infusion of PTH. It is appreciated how the calcemic response is a dynamic situation whose magnitude, among other factors, depends on the degree of renal function (lower calcemic response in rats with more severe renal impairment) and the amount of phosphorus in the diet (lower calcemic response to higher phosphorus content in the diet [and higher serum phosphorus, not represented]) (adapted from Bover et al.58 and Bover et al.59). The term skeletal calcemic response or resistance to PTH has been replaced by hyporesponsiveness to the action of PTH.44](https://static.elsevier.es/multimedia/20132514/0000004100000005/v1_202112230647/S201325142100122X/v1_202112230647/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92lL8PRnKGMnsiVXV6EVJ5QRkjJZIKj2umwUyY+cL6K8cyMCoG0zLrx/ORKa7YTjKb8zGtEpNIMtsJ80ILpYctNh610+ls0qU9EexBTcoHfjwrInf/NwUvNivkFzYe9CcSdZR5I3GrF7+dJvWu95GvSKD7+5tR86XxkOuu5nmeDPLxsvxhqacryftcfN2JG1S0H+2+pBdl27oSYXcpdQ4pYoHOIdX5r9U+Yld+omoNUYeuqSHyVadAglcoJ79qsTvr4Yx94ywNf5qLDR9nyEmWMzEiNZZiaCZJXeVoS2FoeK)
Hyporesponsiveness to the action of parathyroid hormone (PTH). The calcemic response (plasma calcium [Ca] in mg/dl) to the infusion for 48 h of a constant amount of PTH in rats with different degrees of renal function (normal, moderate renal failure and advanced renal failure) and different content is shown of phosphorus in the previous diet (PA: high phosphorus diet [1.2%]; BP: low phosphorus diet [0.3%]; MP: moderate phosphorus diet [0.6%]) rats do not receive calcium in the diet, so the increase in calcium is due to the skeletal response to the infusion of PTH. It is appreciated how the calcemic response is a dynamic situation whose magnitude, among other factors, depends on the degree of renal function (lower calcemic response in rats with more severe renal impairment) and the amount of phosphorus in the diet (lower calcemic response to higher phosphorus content in the diet [and higher serum phosphorus, not represented]) (adapted from Bover et al.58 and Bover et al.59). The term skeletal calcemic response or resistance to PTH has been replaced by hyporesponsiveness to the action of PTH.44
It had been postulated before that the presence of unknown uremic factors, beyond P, could be responsible for the decrease in the calcemic response to PTH in CKD.46 Subsequently, Wills and Jenkins also showed in an in vitro model that serum from uremic patients inhibited the bone resorption induced by PTH, whereas serum obtained after dialysis did not.81 Low molecular weight inhibitors of osteoblast mitogenesis have also been described in uremic plasma,82 and subsequent experimental studies pointed to different uremic toxins83 that trigger oxidative stress, such as indoxyl sulfate (IS) and p-cresyl sulfate84,85 and/or pro-inflammatory oxidized low-density lipoproteins,86 Increased oxidative stress and low-grade inflammation are common in CKD and therefore may cause hyporesponsiveness to the action of PTH, but could also have an indirect effect through other signaling pathways (eg, P, FGF23/klotho, or calcitriol deficiency are associated with oxidative stress and inflammation).44,87In vitro and in vivo studies have shown the possible association of uremic toxins such as indoxyl sulphate (IS) to low bone turnover.88,89 Nii-Kono et al.84 additionally demonstrated that IS induces a state of resistance to PTH by reducing the expression of cAMP and PTH1R induced by PTH, thus reducing the viability of osteoblasts in culture. These authors also demonstrated that the production of free radicals in osteoblasts increased in relation to the concentration of IS added to the medium.84 Furthermore, their results suggested that IS is incorporated into the osteoblasts through an anion transporter, increasing oxidative stress, altering osteoblast function, and regulating PTH1R84 expression. However, correlation of IS levels with bone histomorphometry in CKD patients are contradictory.90 In addition, it has been described that some uremic toxins (for example uric acid) could indirectly stimulate PTH secretion by decreasing the synthesis of calcitriol and the binding to the vitamin D response elements in DNA,91 inducing resistance to the action of another hormone such as calcitriol. In a very recent review.92 the role of uremic toxins on different signaling pathways is expanded, it is described aberrant interorganic relationships and shown how different uremic toxins can interfere with metabolic pathways and the function of multiple molecules and transporters.92
Abnormal metabolism of parathyroid hormone fragments in chronic kidney diseaseThe increased secretion of PTH is mainly responsible for the elevated plasma levels of PTH in CKD. However, kidneys play an important role in its degradation and, in CKD patients, the clearance of PTH is reduced, as is that of other peptide hormones (eg insulin).
More specific reviews discuss extensively how CKD affects the secretion, pulsatility, metabolism, signaling pathways and degradation of PTH [its amino- and carboxy-terminal fragments, also present in other molecules (e.g FGF23)],32,44 as well as how the uremic condition makes so difficult to define the optimal levels of PTH for the different stages of CKD.3
Finally, it is important to highlight that the carboxy-terminal fragments of PTH may not only be biologically active but also, by occupying PTHR1, they antagonize the effects of PTH in kidney and bone.93 In CKD patients, high circulating levels of these PTH antagonizing fragments (also detected by the common “intact” PTH assay) would explain the need for higher PTH levels to prevent ABD. At a certain point, it was described as another mechanism to explain skeletal “resistance” to the action of PTH in uremia.93
In addition to the “classical” PTH1R, it is now accepted that a carboxy-terminal PTHR (PTH4R or PTHR-C) could mediate some unexpected actions of PTH.94,95 Other PTH receptors (PTH2R and PTH3R) have been also described, but their effects in humans and CKD are only partially known.96
Parathyroid hormone signaling cascade, local and systemic factorsAs it has been previously mentioned, PTH exerts its action by binding to PTHR1, expressed mainly in osteoblasts and osteocytes as well as in the kidney.44 Such binding triggers a signal transduction cascade through various pathways (i.e. protein kinases A and C, Wnt/ß-catenin, etc.) that will promote different cellular responses. It is possible that the effects resulting from downstream PTH signaling are counteracted by other competing inhibitory endocrine or paracrine signals induced by other molecules such as FGF23, klotho, sclerostin (antagonist of Wnt/ß-catenin pathway), calcitonin, osteoprotegerin, bone morphogenetic proteins, among others, and together with different local effects such as inflammation, cytokines, oxidative stress, acid-base disorders, or the concentrations of Ca, P, magnesium, fluorine or aluminum.33,44 In fact, the recombinant human protein α-klotho interacts with PTH1R, inhibiting the binding of human PTH to renal tubular cells,97 which inhibited PTH-induced 1α−hydroxylase expression at this level. These results suggested that α-klotho could mediate FGF23 calcitriol synthesis inhibition by blocking PTH signaling.97 Another complex interaction is the effect of butyrate on bone-intestine communication. Butyrate is originated from the intestinal microbiota, and it is known that the depletion of the microbiota decreases the levels of butyrate.98 It has been shown that the restoration of physiological levels of butyrate restored the anabolic action of PTH through the activation of the T cell-dependent Wnt/ß-catenin osteogenic pathway.98
It is very important to highlight the potential role of antagonists of the Wnt/ß-catenin pathway, since PTH is an agonist of this pathway.99 Sclerostin, produced mainly by osteocytes (that also produce FGF23 or osteocalcin), was originally considered a non-classical antagonist of bone morphogenetic proteins.100 Currently, sclerostin is being identified as a soluble inhibitor of the Wnt/ß-catenin pathway through the binding to the LRP5/6 receptors,101,102 resulting in a decrease of bone formation via inhibition of osteoblastgenesis. Furthermore, it appears to play a role as a mediator of other systemic and local factors such as calcitriol, PTH, glucocorticoids, and tumor necrosis factor-α (TNFα).103 Nevertheless, the extent to which circulating levels of sclerostin reflect its bone expression or how it affects other local signaling pathways is still a matter of debate.104 In fact, the expression of sclerostin is increased in patients with incipient CKD, despite having normal levels of PTH,105 and this increase persists in dialysis patients, although to a lesser extent despite having high levels of PTH.106 It is possible that in a near future, the ratio between agonists (PTH) and antagonists (sclerostin, Dkk1 or other associated molecules of the Wnt/ß-catenin pathway) may allow to increase the understanding on the causes and mechanisms of hyporesponsiveness to PTH in CKD and/or even improve the non-invasive diagnosis of bone turnover.
Circulating levels of sclerostin increase with age and with the decrease in renal function,103,107,108 and they are also increased in diabetic patients, regardless of age or sex.109 Sclerostin and its related molecules, such as Dkk1 or serum frizzled 4, have been postulated as possible mediators in the development of ABD33,85 and skeletal resistance to the action of PTH.33,44,85 Thus, it has been postulated that the early inhibition of the osteocyte Wnt/ß-catenin pathway due to the increase of these factors in CKD could be an initial step in the development of renal osteodystrophy, and could explain the high prevalence of low-turnover bone disease (ABD) in early stages of CKD.33,110–112 With CKD progression, the elevation of circulating levels of PTH would eventually overcome the skeletal resistance to PTH generating the classical feature of osteitis fibrosa.8,33 It is not known whether FGF23 and/or α-klotho exert a direct role in this theoretical transition from low to high bone remodeling or whether they would play a role only indirectly through the regulation of PTH secretion.33 In any case, osteocyte dysfunction has already been described in the initial course of CKD.33 Interestingly, the use of anti-sclerostin antibodies (romosozumab) in rat models of progressive renal osteodystrophy appeared to increase the total volume of trabecular bone and the mineralized bone trabecular surface only in animals with low levels of PTH.113 Likewise, bone volume, cortical geometry, and bone architecture improved only if PTH levels were kept relatively low.113 It remains to be clarified whether elevated sclerostin levels are the cause or consequence of the hyporesponsiveness to PTH in CKD.114
From the molecular point of view, it should also be mentioned the potential role of calcitonin (produced by the C cells of the thyroid gland) in the pathogenesis of SHPT and the reduced response to PTH in CKD.115–120 It is also relevant the role of elevated levels of osteoprotegerin, which could favor skeletal resistance to PTH through suppression of the genesis of osteoclasts.121 It is also necessary to remember, as previously mentioned, that different uremic toxins per se have been associated with hyporesponsiveness to PTH in CKD, perhaps by interfering different metabolic pathways, modulating kinase activity (phosphorylation) and/or, binding to second messengers which interfere with intracellular pathways that may affect transcription, protein trafficking and the molecular interactions.91,92
Finally, in the assessment of hyporesponsiveness to PTH in CKD, it should also be taken into consideration other factors such as race, gender,122–125 advanced age or the increasing prevalence of diabetes (known cause of ABD), in addition to the bone turnover itself, or the excessive control of PTH (erroneous normalization of the PTH levels in non-dialysis patients with CKD).
Clinical implications of parathyroid hormone hyporesponsivenessThe skeletal or calcemic resistance to PTH was initially suggested as a mechanism involved in the pathogenesis of SHPT in CKD. With the progression of CKD increasingly higher levels of PTH are necessary to normalize serum calcium concentration, the basis of the trade-off hypothesis.30,50,51 Interest in the concept of resistance to PTH has reemerged with the potential drawback of an excessive suppression of PTH with the use different therapies (Ca-based-P-binders, vitamin D, calcimimetics and PTX) and the observed increase in the prevalence of ABD that is not free of risks, as previously mentioned.15,20–22,44 It has also been recently described that the same PTH levels achieved with different therapies (eg calcimimetics vs. vitamin D) could have different clinical significance due to the different direct impact of these drug on bone independently of their effect on PTH.126,127
It has been said that the term “hyporesponsiveness” to PTH or “relative hypoparathyroidism” is more precise than skeletal resistance to PTH, since the response to PTH is mitigated but not completely absent. London et al., analyzed the intercommunication between bone and vessels128 and showed an association between vascular calcification with reduced PTH levels, a decreased number of osteoclasts, a smaller osteoblast surface and a reduced or absent double tetracycline labeling, although in these cases they also observed a high percentage of aluminum contamination.129 In a cross-sectional study, these authors also found an association between peripheral arterial disease and a significant reduction in the anabolic skeletal response to PTH.130
Additional evidence that hyporesponsiveness to PTH is an important factor in the development of SHPT and/or ABD is obtained from clinical studies that have shown that elevated levels of circulating PTH are necessary to maintain normal bone remodeling.16,131,132 As such, current guidelines3,133,134 suggest that in dialysis patients treatment should be modified to maintain PTH levels at least 2 times higher than the upper limit of normality in order to avoid ABD (more reliable if combined with bone alkaline phosphatase). Likewise, CKD patients prior to dialysis would require higher levels of PTH to maintain a normal osteoblastic surface16 suggesting that dialysis could improve the response to PTH. In fact, this was demonstrated by Torres et al.16 in this study performed using bone biopsies without significant aluminum staining. However, reversibility after dialysis is not a uniform observation.46 Finally, the involvement of so many factors with a complex interrelationship affecting hyporesponsiveness to PTH could explain why there is not a clear direct correlation between the levels of circulating PTH and its clinical consequences, nor with mortality in CKD patients (usually mathematically complex U-, J- or inverted-J shaped) in contrast with the linear associations observed in primary hyperparathyroidism or with serum alkaline phosphatase levels.135
CorollaryBased on the clinical and experimental observations described, hyporesponsiveness to PTH is an integral component of SHPT, the CKD-MBD complex, as the increased levels of PTH are (Fig. 3). The differences observed in the calcemic response to PTH in patients or animals with CKD and normal controls cannot be only justified by the presence of different inactive or antagonistic fragments of PTH, since all individuals and experimental animals received a constant same amount of PTH with uneven results (Fig. 2). P retention, calcitriol deficiency, FGF23/klotho, sclerostin, and/or other uremic factors probably play a role in desensitizing and/or downregulating the PTH receptor or altering subsequent signal transduction pathways.
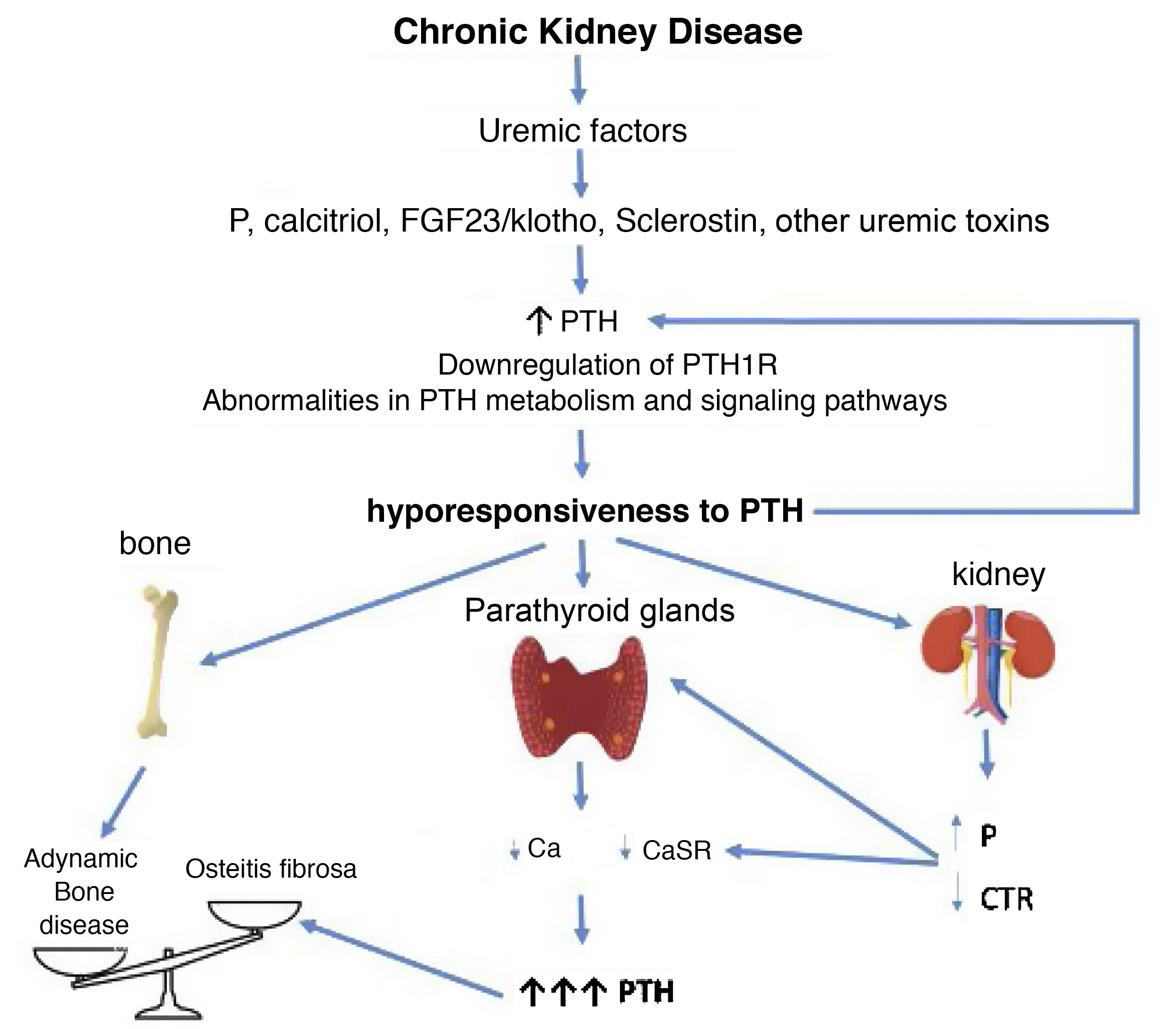
Potential time sequence and consequences of hyporesponsiveness to PTH in CKD. With the development of chronic kidney disease (ie decreased glomerular filtration), factors related to the loss of kidney function (for example the classic phosphorus overload and/or decreased calcitriol) together with factors (known or yet unknown) associated with "uremia" itself, would lead to an increase in PTH. These, together with alterations in PTH metabolism, interferences in its signaling pathways and/or downregulation of its receptors, would condition additional increases in PTH due to its abnormally diminished response. By different mechanisms, the less calcemic response to the action of PTH, the more hormone is necessary to restore calcium levels to normal (greater synthesis and secretion of PTH, as well as greater need for cell proliferation). Likewise, new vicious cycles would be created at the kidney level and imbalances in the bone depending on whether anabolic or inhibitory elements prevail. Osteitis fibrosa is the bone expression of secondary hyperparathyroidism (high turnover bone disease) and adynamic bone disease is the result of low bone turnover.
CTR: calcitriol; FGF-23: Fibroblast Growth Factor 23; P: phosphorus; PTH1R: parathyroid hormone receptor (PTH); CaSR: calcium sensing receptor (adapted from Bover et al.148).
Although skeletal resistance to PTH was initially suggested as a mechanism involved in the pathogenesis of SHPT in CKD, the “hyporesponsiveness” to PTH has also been associated with an increased prevalence of ABD (Fig. 3) which could also be explained by the increased number of elderly and diabetic patients with CKD, and the excessively aggressive treatments to reduce PTH. Therefore, as nephrologists we should take into account this already classic concept of hyporesponsiveness to PTH when establishing strategies to treat SHPT in our patients. Consequently, it is important to avoid complete normalization of PTH levels, as it is suggested in current guidelines.3,134 Conversely, a progressive and persistent increase in PTH levels will transform an initially adaptive clinical situation into a clearly maladaptive one.
Defining an optimal individual PTH target is currently impossible, but it could be achieved at the population level.44 It is a matter of debate whether it is more appropriate to wait for the development of severe SHPT before starting treatment, as suggested by the latest KDIGO3 guidelines, or to simply avoid a normalization of PTH levels, as suggested by others.134 In any case, there seems to be a certain consensus that the minimum PTH level at the initiation of regular dialysis should be approximately 2 times the upper limit of normal for the kit used.3,134 In fact, we could now speculate that if the aforementioned factors (retention of P, hypovitaminosis D, down-regulation of PTHR1, uremic toxins, etc.) were corrected, the bone and renal tubule might respond normally to normal PTH levels, and PTH would not have to rise maladaptively.
It is probably not plausible to propose a single recommendation for the entire population of CKD patients about PTH levels (neither before nor after starting dialysis) and we should advice a more personalized patient management (probably sharing therapeutic decisions with the patient),25,136,137 since other factors such as age, presence or absence of diabetes, metabolic syndrome, nephrotic syndrome, vascular calcification, risk of fracture (recently incorporated into our clinical practice) or other biochemical markers (eg alkaline phosphatase should be considered). Other factors and concepts recently identified that require future research (uremic toxins, FGF23/klotho, Wnt/β-catenin, activin A receptor type 2 activation pathways, ‘osteocytic osteolysis’, etc.) are likely to provide new insights.138
Today, there is no biomarker better than PTH despite its limitations, and we certainly are in need for studies to improve diagnostic precision. More frequent monitoring to identify trends in PTH levels seems the appropriate way to proceed until new and better molecular targets and treatments demonstrate more efficacy than PTH in clinical practice.
Finally, resistance to the biological action of other hormones, such as insulin, calcitriol, growth hormone, and FGF23 are well recognized in CKD,139–142 as well as the decreased expression of various receptors (i.e. VDR, CaSR, FGFR/klotho).65,143–147 In fact, in the same way that uremia is a complex metabolic disorder that involves numerous molecules, metabolic alterations, and aberrant systemic signaling pathways,92 uremia could also be considered as a disease that, through unknown mechanisms, widely affects the functionality of different types of receptors (uremia, a “receptor disease”).44,148 Thus, studies are needed to discover new therapies that could be useful even in CKD even beyond hyporesponsiveness to PTH.
ConclusionSHPT is one of the integral components of the CKD-MBD complex and hyporesponsiveness (or resistance) to PTH is a mechanism that contributes to its generation. The hyporesponsiveness to PTH is the result of multiple complex mechanisms (many of them have not been identified yet) and are not exclusive to this hormone. As a consequence, a certain degree of SHPT is a necessary adaptive mechanism in patients with CKD. Only future studies at the molecular level will be able to answer the in-depth mechanisms of hormonal hyporesponsiveness in CKD, an essential step toward the discovery of useful therapeutic strategies beyond hyporesponsiveness to PTH.
Key concepts- 1
SHPT is one of the integral components of the complex CKD-mineral and bone disorders (CKD-MBD).
- 2
PTH is considered a “uremic toxin” with systemic deleterious effects beyond the bone.
- 3
Hyporesponsiveness (or resistance) to the action of PTH refers to the decreased response to the action of PTH (i.e. calcemic response) that is observed from early stages of CKD.
- 4
Today the term “hyporesponsiveness” is preferred to that of “resistance” to the action of PTH, since there is a decrease rather than an absence of response to PTH in CKD.
- 5
Hyporesponsiveness to the action of PTH is part of the pathogenic mechanisms of SHPT in CKD.
- 6
There are multiple mechanisms associated with hyporesponsiveness to PTH, such as decreased levels of calcitriol, P retention, under-regulation of renal and bone PTHR1, accumulation of uremic toxins and different molecules such as αKlotho, FGF-23, sclerostin or various factors converging in downstream signaling pathways.
- 7
It is not possible to identify an optimal PTH target for all patients with CKD, so individualized management may need to be implemented.
- 8
No attempt should be made to maintain normal PTH values in CKD patients, since a certain degree of SHPT is a necessary adaptive mechanism.
- 9
Clinical practice guidelines emphasize the importance of treating trends (considering PTH, Ca, and P as a whole) and not reacting to isolated PTH determinations.
- 10
Future studies at the molecular level could provide an answer to the intimate mechanisms of hyporesponsiveness to PTH in CKD and to the functional impairment of different receptors in uremia (“uremia, a receptor disease”).
The authors have no conflict of interest directly related to this topic. J. Bover has received conference and consulting fees from Abbvie, AMGEN, Sanofi, and VIFOR-Fresenius-Renal Pharma. P. Ureña has received conference and consulting fees from AMGEN, Astellas, GSK, Hemotech, Leo-Pharma, Sanofi, and VIFOR-Fresenius-Renal Pharma. A. Martín-Malo has received conference and consulting fees from VIFOR-Fresenius-Renal Pharma, Medtronic, and AstraZeneca. M Rodriguez has received conference and consulting fees from AMGEN, Viphor and Kiowa Kirin.
ThanksJ. Bover belongs to the “RedinRen National Network” (RD06/0016/0001 and RD12/0021/0033) and the “Spanish National Biobank Network” (RD09/0076/00064), as well as to the “Catalan Research Group” AGAUR (2009 SGR-1116). A. Torres, A Martin Malo and M Rodriguez also belongs to the “RedinRen National Network” (RD16/0009/0031). We thank Mr. Ricardo Pellejero for his invaluable, constant and enthusiastic bibliographic assistance and Ms. María Fernanda Coll for her help in graphic design.
Please cite this article as: Bover J, Arana C, Ureña P, Torres A, Martín-Malo A, Fayos L, et al. Hiporrespuesta o resistencia a la acción de la hormona paratiroidea en la enfermedad renal crónica. Nefrologia. 2021;41:514–528.



