

Cystinosis is an inborn error of metabolism, clinically characterised by severe renal involvement and development of corneal cystine deposits, especially in the adult form of the disease. Cystinosis is a treatable condition. Therefore, an early diagnosis is necessary to start therapy. For biochemical confirmation of the condition it is necessary to quantify intracellular cystine concentrations. For this, different methods have been described with variations in cell isolation strategies and the amino acid quantification techniques used. In order to improve confirmatory biochemical diagnosis in our setting, a protocol for intraleukocitary cystine quantification was established.
MethodsA high performance liquid chromatography based method for cystine quantification in polymorphonuclear cells was implemented. Evaluation of the best anticoagulant to use and temperature stability of the sample at 4°C were performed. In addition, we established reference values for our population.
ResultsIt was determined that intraleukocitary cystine quantification must be performed in blood samples containing acid-citrate-dextrose (ACD) as anticoagulant. Samples must be processed immediately due to their poor stability even when refrigerated. Based on the results from 50 healthy individuals, the cut-off point established for our population was 0.34nmol1/2/cystine/mg.
ConclusionThe adaptation performed to the cystine quantification method here presented the highest control population that has been reported in the literature so far. Our results highlight the need for making available a cystine quantification method locally and confirm the convenience for each laboratory to establish its own reference values to provide greater reliability for interpreting results.
La cistinosis es un error innato del metabolismo cuyas características clínicas incluyen compromiso renal severo, formación de cristales de cistina en la córnea, especialmente en la presentación adulta de la enfermedad. Es una enfermedad tratable por lo cual establecer el diagnostico de forma oportuna es fundamental para iniciar terapia. Para la confirmación bioquímica de la enfermedad se requiere determinar las concentraciones intracelulares de cistina, para lo cual se han reportado diferentes métodos tanto para el aislamiento de las células como las técnicas de cuantificación del aminoácido. Con el objetivo de mejorar el diagnóstico bioquímico confirmatorio en nuestro medio establecimos un protocolo de cuantificación intraleucocitaria de cistina.
MétodosSe realizó implementación de un método de cuantificación de cistina en polimorfonucleares por cromatografía líquida de alta resolución, evaluando el mejor anticoagulante a utilizar, la estabilidad de la muestra a 4°C y estableciendo valores de referencia para nuestra población.
ResultadosSe determinó que la muestra para cuantificación intraleucocitaria de cistina debe ser anticoagulada mediante adición de ácido Cítrico-Dextrosa (ACD) como anticoagulante. La muestra debe ser procesada inmediatamente dada su baja estabilidad incluso en refrigeración. Con 50 individuos sanos se estableció como punto de corte para nuestra población 0.34nmol1/2cistina/mg.
ConclusiónLa adaptación realizada del método de cuantificación de cistina utiliza el número más alto de muestras control hasta ahora reportado en la literatura. Nuestros resultados dan cuenta de la necesidad de implementar el método a nivel local y reafirman la conveniencia de que cada laboratorio establezca sus propios valores de referencia para proporcionar una mayor confiabilidad a la hora de interpretar los resultados.
Cystinosis is an innate metabolism error caused by the lysosomal enzyme cystinosin, a cystin/H (+) transporter responsible for transporting cystine from inside the lysosomes to the cell cytoplasm;1 the enzyme is coded by the CTNS gene located on 17p13.2.2 This disease is the most common cause of Fanconi syndrome during the first year of life. Clinically, it can also be presented in a delayed form during childhood or adolescence as moderate Fanconi syndrome or just isolated proteinuria, even into adulthood, and the main manifestation is ocular compromise represented by the formation of cystine crystals which are deposited onto the cornea.2
The wide clinical variability of the disease makes many cases difficult to diagnose and requires a high level of experience and knowledge by health care personnel. Because of this, although it is possible to make an early diagnosis, in many cases it may be delayed, even up to the end stage of kidney failure in the infantile presentations and up to severe ocular compromise in the adult form. In any of the cases, the biochemical findings that accompanies the clinical impression must be the demonstration of cystine accumulation within the lysosome of polymorphonuclear cells.2 Making a fast diagnosis is essential, since there is an effective treatment to deplete intralysosomal cystine, which would have a positive impact on the disease's progression and prognosis.2,3
Once there is a clinical suspicion, quantification of white blood cell cystine makes it possible to confirm the diagnosis of cystinosis, after which a disease-specific treatment can be started using the analogue of the altered protein.3–5 However, this technique is not regularly performed, even in laboratories specialising in metabolic diseases, primarily due to the difficulty in obtaining clear fractions of polymorphonuclear cells.6,7 With the availability of a therapy for this disease,3,4,8,9 there is a need for a rapid and precise diagnosis, to have a positive impact on quality of life and minimising renal compromise.
High performance chromatography has been the traditional method for quantifying amino acids in biological fluids, although in the case of white blood cell cystine, multiple variations of the methods used are found in the literature, both for isolating the cells and quantifying the amino acid.5,10,11 Taking the above into consideration and knowing that the implementation of a specific method may vary between laboratories, it is important to establish specific conditions for each laboratory that wants to perform cystine quantifications, as well as to determine the reference values for each population, to prevent biases in the interpretation. Therefore, this work presents the standardisation of a method to quantify cystine in polymorphonuclear cells using HPLC and establishes reference values for the Colombian population.
MethodologySampleThe sample used was venous blood, obtaining a minimum of 6ml (one tube). No fasting or special conditions were required for collecting the sample. Since the main anticoagulants used in the literature are heparin and acid-citrate-dextrose (ACD),7,12–14 these two anticoagulants were compared using Vacutainer® tubes containing heparin sodium or ACD solution B. Additionally, the stability of the whole blood sample was evaluated when stored at 4°C for 8 and 24h.
Separation of polymorphonuclear cellsA polymorphonuclear isolation protocol from whole blood was standardised based on the one described by Levtchenko et al. and the recommendations of the program by the European Research Network for evaluation and improvement of screening, Diagnosis and treatment of Inherited disorders of Metabolism.7,15 Briefly, the method consists of a sequential separation scheme using first dextran 5% and then Ficoll® 1119, followed by eliminating the red blood cells present in the sample by osmotic lysis. Lastly, the obtained specimen was resuspended in N-ethylmaleimide (Sigma 04259) 5.2mM in distilled water, which must be processed immediately.
Quantification of white blood cell cystineBefore the analysis with chromatography, the obtained cell specimen undergoes ultrasound rupture by applying four 15-second cycles, each at a low intensity (20%). Afterwards, the sample was deproteinized by adding a solution of sulfosalicylic acid 12%. The cystine was quantified via high performance liquid chromatography using a Biochrom 30 amino acid analyser. The quantification was performed using a commercial amino acid solution containing cystine at a concentration of 125 μM (Sigma A6282) as a standard. The control run was performed using N-leucine (Sigma N6877) as an internal standard. The equipment performs the separation by anionic exchange and post-column derivation with ninhydrin, with readings at 537nm. Taking into account that the quantified cystine molecules were released from the white blood cells, the report was given in nmol 1/2 cystine and normalised for the protein concentration of the sample.
Quantification of the proteinThe pellet obtained from deproteinizing the sample was resuspended in NaOH 0.1N and the total protein quantified using the Folin-Lowry method against the bovine serum albumin (Sigma A2153) calibration curve between 0.125 and 3mg/ml.16
Establishment of reference values for the local populationWith the aim of establishing reference values in our population, 50 healthy, adult volunteers were selected by open call with no regard for age or gender. After signing the informed consent form, a blood sample was collected to quantify the white blood cell cystine using the established method. Additionally, the results were validated using samples from four patients with a confirmed diagnosis of cystinosis.
Results and discussionAnticoagulant selectionIn the analyses carried out, it was observed that using ACD as an anticoagulant resulted in better chromatography peak recovery and better phase resolution in the dextran gradient step. Based on these results, ACD was selected as the anticoagulant for developing the quantification process.
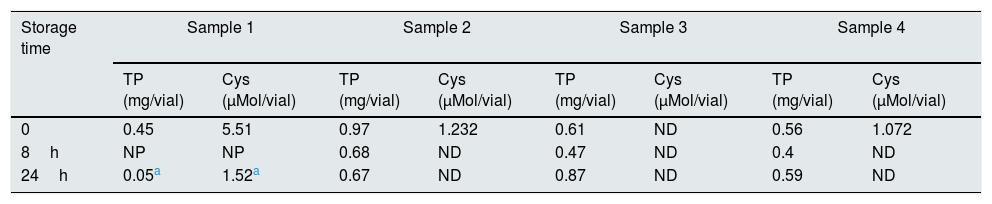
Sample stabilityThe storage stability of the blood sample was evaluated under two storage conditions: 8 and 24h at 4°C. The results show that storing the samples leads to false decreases in cystine, which became undetectable in most cases (Table 1). Only sample 1, which had the highest cystine concentration, show detectable values after 30h of storage.
Results of the stability assays.
| Storage time | Sample 1 | Sample 2 | Sample 3 | Sample 4 | ||||
|---|---|---|---|---|---|---|---|---|
| TP (mg/vial) | Cys (μMol/vial) | TP (mg/vial) | Cys (μMol/vial) | TP (mg/vial) | Cys (μMol/vial) | TP (mg/vial) | Cys (μMol/vial) | |
| 0 | 0.45 | 5.51 | 0.97 | 1.232 | 0.61 | ND | 0.56 | 1.072 |
| 8h | NP | NP | 0.68 | ND | 0.47 | ND | 0.4 | ND |
| 24h | 0.05a | 1.52a | 0.67 | ND | 0.87 | ND | 0.59 | ND |
Cys, cystine; ND, not detectable; NP, not performed; TP, total protein.
Additionally, a loss of quality was observed in some samples, as evidenced by the decrease in the total quantity of protein (Table 1).
The results obtained make it possible to conclude that the whole blood sample is not stable, therefore it should be collected at the place where the process is performed.
Separation of polymorphonuclear cellsTo confirm that polynuclear and mononuclear cells were obtained in the Ficoll step, the cell fractions were quantified by cytometry using Beckman Coulter DxH 800 equipment, which showed that the working fraction for quantifying the polymorphonuclear cell population exceeded 90% of all cells, (91.35–99.44; n=4).
Quantification of cystineUsing the standard HPLC quantification method, commercial cystine standards were processed with concentrations between 0 and 0.5mM, a range which covers the concentration of affected individuals (nmol level). The final calibration curve obtained shows a 99.84% correlation between the concentration of real (prepared) cystine and cystine calculated by HPLC, which validates the ability of HPLC to accurately detect the concentration of cystine present in a sample.
Establishment of reference values for the local populationOnce the cystine concentration of the healthy volunteers was determined, the frequency histogram was established and the 90th percentile was considered to establish a cut-off point of 0.34nmol 1/2 cystine/mg. The observed spread is comparable to that previously reported by Gertsman et al., who worked with a sample of 10 patients14 (Fig. 1A). Additionally, the values reported in different studies range between 0 and 0.2nmol 1/2 cystine/mg protein, approximately, which coincides with the results of around 70% of our population.2,3,6,15 It is important to highlight that the reports in the general literature use populations with a very low number of individuals (between 2 and 30, approximately), which decreases the probability of finding the high values seen in this study and which correspond to only 10% of our population.
 Cystine concentration distribution histogram in normal individuals (n=50). (B) Concentration of white blood cell cystine in a healthy and affected population.")
Once the quantification protocol was established, the white blood cell cystine concentration was determined in the four affected individuals, one individual suspected of being affected, and another heterozygous individual (one of the parents of the patients was included in the study).
As shown in Fig. 1B, while the populations of the healthy population reached up to 0.7nmol 1/2 cystine/mg protein, the values of the patients are above this level: between 1 and 12nmol 1/2 cystine/mg protein. These results are comparable to those observed in other studies.6,7,14,17 It is worth clarifying that the patient whose value was 1 is on treatment. In addition, these values are similar to those reported by other authors for treated patients.7,17
The processed sample from the heterozygous individual showed intermediate values (1.06nmol 1/2 cystine/mg protein), which fall into the established “grey”area according to the previously established reference values. This agrees with what has been reported in the literature, where heterozygous individuals may present values which range between normal and five times higher than the cut-off point, although still lower than those observed in patients (usually more than 10 times higher than the cut-off point). Lastly, the individual being studied for cystinosis had results considered to be normal.
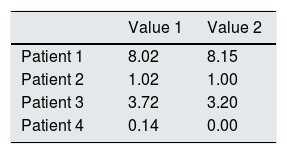
Additionally, the validity of the proposed methodology was checked by simultaneously determining the cystine in the samples processed by us (laboratory 1) and by a reference laboratory commonly used for sending samples from our country (laboratory 2). The results presented in Table 2 show good agreement between laboratories, which validates the method used.
Inter-laboratory reproducibility of white blood cell cystine quantification.
| Value 1 | Value 2 | |
|---|---|---|
| Patient 1 | 8.02 | 8.15 |
| Patient 2 | 1.02 | 1.00 |
| Patient 3 | 3.72 | 3.20 |
| Patient 4 | 0.14 | 0.00 |
Value 1, laboratory 1 quantification; Value 2, laboratory 2 quantification.
All concentrations expressed as nmol 1/2 cystine/mg protein.
Cystinosis is an innate metabolism error and one of the causes of childhood kidney disease which can be life-threatening if not diagnosed and treated on time. For this reason, it is essential to make a quick and precise diagnose, for which the white blood cell cystine quantification method, is needed locally. The adaptation of the HPLC cystine quantification method in polymorphonuclear cells uses the highest number of control samples reported to date in the literature. The protocol established uses a blood volume considered suitable for individuals from an early age; it is important to taken into account that other general laboratory tests, such as immunology and cytometry studies, as well as lactic acid determinations, usually use larger blood volumes than those used by us with this technique. Even so, it would be advisable in future studies to adapt the method to smaller blood volumes, to make it more applicable to younger infants.
Implementing this technique is of great importance for diagnosing cystinosis in Colombia and accounts for the need to implement the method locally, given the sample instability. Additionally, some differences were observed compared to other reported values, reaffirming the advantage of each laboratory establishing its own reference values to provide greater reliability when interpreting the results.
Conflict of interestThe authors have no conflicts of interest to declare.
The authors thank the company Recordati Rare Diseases for its economic support, by funding project ID # 7350, Pontificia Universidad Javeriana, Bogota (Colombia).
Please cite this article as: Guevara Morales JM, Echeverri Peña OY. Implementación de un método para la cuantificación de cistina intraleucocitaria como apoyo diagnóstico para la cistinosis. Nefrologia. 2020;40:99–103.



