



There are many experimental data supporting the involvement of aldosterone and mineralcorticoid receptor (MR) activation in the genesis and progression of chronic kidney disease (CKD) and cardiovascular damage.
Many studies have shown that in diabetic and non-diabetic CKD, blocking the renin-angiotensin-aldosterone (RAAS) system with conversion enzyme inhibitors (ACEi) or angiotensin II receptor blockers (ARBs) decreases proteinuria, progression of CKD and mortality, but there is still a significant residual risk of developing these events.
In subjects treated with ACEi or ARBs there may be an aldosterone breakthrough whose prevalence in subjects with CKD can reach 50%. Several studies have shown that in CKD, the aldosterone antagonists (spironolactone, eplerenone) added to ACEi or ARBs, reduce proteinuria, but increase the risk of hyperkalemia. Other studies in subjects treated with dialysis suggest a possible beneficial effect of antialdosteronic drugs on CV events and mortality. Newer potassium binders drugs can prevent/decrease hyperkalemia induced by RAAS blockade, and may reduce the high discontinuation rates or dose reduction of RAAS-blockers.
The nonsteroidal MR blockers, with more potency and selectivity than the classic ones, reduce proteinuria and have a lower risk of hyperkalemia. Several clinical trials, currently underway, will determine the effect of classic MR blockers on CV events and mortality in subjects with stage 3b CKD and in dialysis patients, and whether in patients with type 2 diabetes mellitus and CKD, optimally treated and with high risk of CV and kidney events, the addition of finerenone to their treatment produces cardiorenal benefits.
Large randomized trials have shown that sodium glucose type 2 cotransporter inhibitors (SGLT2i) reduce mortality and the development and progression of diabetic and nondiabetic CKD. There are pathophysiological arguments, which raise the possibility that the triple combination ACEi or ARBs, SGLT2i and aldosterone antagonist provide additional renal and cardiovascular protection.
Hay abundantes datos experimentales que sustentan la participación de la aldosterona y la estimulación del receptor mineralcorticoide (RM) en la génesis y progresión de la enfermedad renal crónica (ERC) y en el daño cardiovascular.
Muchos estudios han demostrado que en la ERC diabética y no diabética, el bloqueo del sistema renina angiotensina-aldosterona (SRAA) con inhibidores de enzima de conversión (iECA) ó antagonistas del receptor de angiotensina II (ARA2) disminuye la proteinuria, la progresión de la ERC y la mortalidad, pero persiste todavía importante riesgo residual de desarrollo de estos eventos.
En sujetos tratados con iECA ó ARA2 puede haber un escape de la aldosterona cuya prevalencia en sujetos con ERC puede alcanzar el 50 %. Diversos estudios han demostrado que, en la ERC, los fármacos antialdosterónicos clásicos (espironolactona, eplerenona) añadidos a iECA ó ARA2, reducen la proteinuria, pero aumentan el riesgo de hiperkaliemia. Otros estudios en sujetos tratados con diálisis sugieren un posible efecto beneficioso de los antialadosterónicos sobre eventos CV y mortalidad. Los nuevos ligadores intestinales de K+ pueden prevenir ó reducir la hiperkaliemia inducida por el bloqueo del SRAA, y disminuir la desprescripción o la reducción de dosis de fármacos bloqueantes del SRAA.
Los bloqueantes del RM no esteroideos, con más potencia y selectividad que los clásicos, reducen la proteinuria y tienen menos riesgo de hiperkaliemia. Varios ensayos clínicos, actualmente en realización, determinarán el efecto de los bloqueantes clásicos del RM sobre eventos CV y mortalidad en sujetos con ERC estadio 3b y en enfermos en diálisis, y si en enfermos con diabetes mellitus tipo 2 y ERC, óptimamente tratados y con elevado riesgo de eventos CV y renales, la adición de finerenona a su tratamiento produce beneficios cardiorrenales.
Los inhibidores del cotransportador sodio-glucosa tipo 2 (iSGLT2) han demos-trado reducir la mortalidad y el desarrollo y progresión de la nefropatía diabética y no diabética. Hay argumentos fisiopatológicos, que suscitan la posibilidad de que la triple combinación, iECA ó ARA2, iSGLT2 y antagonista de aldosterona ofrezca mayor protección renal y vascular.
Considerable evidence implicates the renin-angiotensin-aldosterone system (RAAS) in the progression of chronic kidney disease (CKD) Blocking the generation and action of angiotensin II (AII) by AII converting enzyme inhibitors (iACEi) and the A II AT1 receptor blockers (ARA2) have shown to be useful to reduce the progression of diabetic and non-diabetic nephropathy1–5 and to decrease cardiovascular (CV) morbidity and mortality and mortality of any cause.6 Mineralocorticoid receptor (MR) blockade with spironolactone and eplerenone adds an antiproteinuric effect when associated to ACEI or ARA2. However, so far, there is no consistent evidence show in a benefit of MR blockade in the progression of CKD or in reducing CV events and total mortality in CKD. However, MR blockade in patients with heart failure with reduced ejection fraction (EF) decrases CV events and mortality.7–9
Physiology/pathophysiology of aldosterone/MRAldosterone was isolated in 1953 by Simpson et al.10 It is produced mainly by glomerulosa cells of the adrenal glands. Under certain circumstances, aldosterone is produced in the heart, blood vessels, and adipocytes.11,12 Increased expression of aldosterone synthetase (AS) has been shown in human coronary arteries from multi-organ donors, and it is increased in subjects with renal failure and correlates with the vascular expression of the osteoblastic transforming factor: core binding factor alpha 1 (CBFa1).13
The A II and an increase in K+ concentration are the main aldosterone secretagogues. However, other factors such as ACTH, catecholamines, endothelin, and adipocyte-derived products, such as leptin, adiponectin, and C1qTNF-related protein 1 (CTRP1), also stimulate adrenal secretion of aldosterone.12,14
In culture of aortic vascular smooth muscle cells, the presence of phosphorus in the medium induces an increase in the vascular expression of AS.13 This fact may be relevant in CKD. In addition, in adrenal glands it has been demonstrated colocalization of klotho-AS and in klotho haplodeficient animals there is an increase in the expression of AS.15
The MR was cloned in 1997.16 It is a protein of 107 kD encoded by a gene located on chromosome 14. It has three domains: an N-terminal domain, another domain that binds to DNA and a third domain that is assembled to the ligand. Certain coactivators and co-repressors bind some domains, that can be tissue-specific and modulate specific actions in certain organs.17 There are MR in the renal collecting duct and colon (classic sites), and also in other locations18–25 (Table 1).
Cortisol also binds to MR. In epithelial cells and vascular endothelium, the enzyme 11-βOH-steroid dehydrogenase converts cortisol into cortisone, thus preventing its binding to MR.
The MR is not inserted directly into the cell membrane. In a basal state, not bound to the ligand, the RM is located predominantly in the cytoplasm bound to chaperones that facilitate its binding to the ligand and serve as shuttles of the RM-ligand complex to the nucleus.25 The aldosterone-RM binding promotes several effects: (1) the RM complex and its ligand are translocated to the nucleus where it forms dimers and activates genes encoding certain proteins (genomic effects) (Fig. 1); There may be interaction with other cytoplasmic transcription factors such as nuclear factor of activated T-cells (NAFT) and cAMP response element-binding protein (CREB) that can modify genomic signals; (2) in addition to the genomic effects, there may be fast non-genomic effects. The aldosterone-MR complex can directly activate cytoplasmic kinases through its interaction with membrane receptors.25,26 Aldosterone binds to MR that is anchored in the membrane by caveolin and striatin and interacts with membrane receptors such as epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR), insulin like growth factor receptor (IGF1R), vascular endothelial growth factor receptor (VEGFR), G protein coupled estrogen receptor (GPER) and angiotensin II receptor type I (AT1R). This interaction is responsible for the non-genomic effects of aldosterone mainly related with electrolyte transport and vasomotor tone (Fig. 1).
 is predominantly located in the cytoplasm, forming part of a complex with the chaperone heat shock protein (Hsp90), which facilitates its binding to the ligand, and with the immunophilin FK506-binding protein of 51 kDa (FKBP51). After binding to the ligand, immunophilin FKBP51 is replaced by FBBP52, which facilitates the transit of the RM-ligand complex from the cytoplasm to the nucleus. Once in the nucleoplasm, the RM-ligand dissociates from the chaperone and immunophilin to bind to DNA and form dimers. MR binds to specific DNA sequences, the hormone response element (HRE) to regulate the transcription of target genes (some of these are shown in the figure). MR-mediated transcriptional activity can be increased or repressed by regulatory proteins such as steroid receptor coactivator 1 (SRC1), cAMP-response element binding protein (CBP), and transcriptional intermediary factor 1a (TIF1a), among others. 2. The RM-ligand complex can interact with other transcription factors such as cAMP response element-binding protein (CREB) and nuclear factor of activated T-cells (NFAT), which can influence transcription. Aldosterone also binds to MR anchored in the cell membrane by caveolin (Cav1) and striatin, transactivating membrane receptors such as epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR), insulin like growth factor receptor (IGF1R), vascular endothelial growth factor receptor (VEGFR), G protein coupled estrogen receptor (GPER) and angiotensin II receptor type I (AT1R). All this promotes signals that involve, among other effects, kinase activation. These events are responsible for the rapid, non-genomic effects of aldosterone. 3. Activation of the RM receptor is possible without the ligand. The activation of Ras-related C3 botulinum toxin substrate 1 (Rac1) by stimuli such as oxidative stress, glucose, AII, among others, can promote the translocation of the RM to the nucleus and trigger its transcriptional activity.")
At a basal state, the mineralocorticoid receptor (MR) is predominantly located in the cytoplasm, forming part of a complex with the chaperone heat shock protein (Hsp90), which facilitates its binding to the ligand, and with the immunophilin FK506-binding protein of 51 kDa (FKBP51). After binding to the ligand, immunophilin FKBP51 is replaced by FBBP52, which facilitates the transit of the RM-ligand complex from the cytoplasm to the nucleus. Once in the nucleoplasm, the RM-ligand dissociates from the chaperone and immunophilin to bind to DNA and form dimers. MR binds to specific DNA sequences, the hormone response element (HRE) to regulate the transcription of target genes (some of these are shown in the figure). MR-mediated transcriptional activity can be increased or repressed by regulatory proteins such as steroid receptor coactivator 1 (SRC1), cAMP-response element binding protein (CBP), and transcriptional intermediary factor 1a (TIF1a), among others. 2. The RM-ligand complex can interact with other transcription factors such as cAMP response element-binding protein (CREB) and nuclear factor of activated T-cells (NFAT), which can influence transcription. Aldosterone also binds to MR anchored in the cell membrane by caveolin (Cav1) and striatin, transactivating membrane receptors such as epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR), insulin like growth factor receptor (IGF1R), vascular endothelial growth factor receptor (VEGFR), G protein coupled estrogen receptor (GPER) and angiotensin II receptor type I (AT1R). All this promotes signals that involve, among other effects, kinase activation. These events are responsible for the rapid, non-genomic effects of aldosterone. 3. Activation of the RM receptor is possible without the ligand. The activation of Ras-related C3 botulinum toxin substrate 1 (Rac1) by stimuli such as oxidative stress, glucose, AII, among others, can promote the translocation of the RM to the nucleus and trigger its transcriptional activity.
The genomic signals of MR-aldosterone can be modified by epigenetic signals and undergo post-transcriptional changes. Also the redox status and the amount of reactive oxygen species (ROS) influence the cellular processes and even the activation mechanism triggered by the activation of MR.
The activation and transcriptional effect of MR may occur without its binding to the ligand (aldosterone or cortisol). Activation of ras-related C3 botulinum substrate 1 (Rac1) may cause MR stimulation with its transcriptional activity independently of aldosterone27 (Fig. 1). Rac1 is a member of the RhoGTPase subfamily that transduces extracellular signals from G protein-coupled receptors, integrins, and growth factors to effector molecules that modulate multiple signaling pathways. Various stimuli, such as mechanical stretching, inflammatory cytokines, growth factors, glucose, aldosterone, and oxidative stress, among others, activate Rac1.28 In vitro studies show that Rac1 improves the transcriptional activity of MR by increasing its nuclear translocation.29 Studies in animal models with increased Rac1 activation, the Rac1-MR interaction is associated with proteinuria and podocyte damage, without changes in aldosterone levels. Pharmacological inhibition of Rac or the use of MR antagonist inhibits the hyperactivity of MR and reduced renal histological changes and proteinuria.29
Mesangial cells cultured in high glucose increase Rac1 activity, and mayenhance the transcriptional activity of the MR induced by aldosterone; also it may increase transcriptional activity of the RM in the absence of aldosterone in the medium.30 In animal models of obesity-related diabetic nephropathy in which there is an increase in plasma aldosterone concentration and renal Rac1 activity, administration of a specific Rac inhibitor suppressed renal Rac1, decreased MR activation, and attenuated renal damage, all without changes in the aldosterone level.30 In the murine model of salt-sensitive HT, the high-salt diet increases renal Rac1 activity and produces MR activation at the renal level, and induces HTN and kidney damage despite low plasma aldosterone levels. The opposed, Rac inhibition prevented hypertension and kidney damage. The infusion of aldosterone enhanced the activation of the MR by Rac1 which demonstrate an additive effect of Rac1 and aldosterone.31
In most of these studies, the increase in MR transcriptional activity induced by Rac1 was due to an increase in nuclear translocation of MR. There are other mechanisms could participate in the increase in MR signals mediated by Rac1: both the amount and modifications of signals (phosphorylation, acetylation, sumoylation (small ubiquitin-related modifier) of MR, changes in its efficacy after activation due to epigenetic modifications or recruitment of co-regulators, among others.28
Aldosterone-independent MR modulation by Rac1 may be of great relevance not only because it enhances the MR activation exerted by aldosterone, but also because it activates MR, promoting organ dysfunction in situations in which the plasma concentration of aldosterone is normal or reduced, as frequently occurs in DM with renal involvement and in salt-sensitive HT, among others. It would also explain that the effect of MR antagonists is not correlated on many occasions with the plasma concentration of aldosterone.
Effects of aldosterone on the renal tubuleThe first action described of aldosterone was on the main cells of the renal collecting duct. After translocation to the nucleus, the aldosterone-MR complex interacts with a large number of genes that encode proteins such as the alpha subunit of the epithelial sodium channel (ENaC) and serum and gluco-corticoid-regulated kinase 1 (SGK1), which control the amount and the activity of ENaC in the apical part of the membrane and of the Na-K-ATPase pump in the basolateral membrane. The electrogenic increase in tubular reabsorption of Na+ secondary to the activation of ENaC favors the secretion of K+ by the renal outer medullary K+ channel (ROMK).32 Recent findings show that in the presence of hypokalemia, aldosterone activates the pendrin, ATPase dependent H+ secretion and the thiazide sensitive cotransporter Na+–Cl− (NCC).33–35
Aldosterone exerts tubular effects that affect the afferent arteriole and, therefore, glomerular filtration (GFR). The macula densa (MD) contains MR whose activation by aldosterone increases nitric oxide production with attenuation of the tubulo-glomerular feedback (TGF) response (decrease in the afferent arteriole vasoconstrictor response induced by the delivery of Na+ to MD).36 In addition, by activating ENaC and increasing distal Na+ reabsorption, it induces vasodilation of the afferent arteriole (tubule connecting-glomerulus feedback [TCGF]), which also antagonizes TGF.37,38
Vascular effects of aldosteroneIn addition to this indirect effect on glomerular hemodynamics, aldosterone has a direct impact on the systemic and renal vasculature. There are MR on the endothelium and the smooth muscle cell. In blood vessels, it regulates genes involved in fibrosis, calcification, and inflammation. The degree of regulation depends on other factors, such as type of flow (laminar/turbulent) and oxidative stress, among others. Endothelial MR activation produces an increase in endothelial ENaC expression, intensifies oxidative stress and inflammation, contributing to endothelial dysfunction and increased arterial stiffness.39,40
In the vascular smooth muscle cell, MR activation contributes to the regulation of vascular tone through a series of events (activation Src kinase, Rho kinase and placental growth factor [PlGF], among others) that induce proliferation, fibrosis, remodeling and increase of arterial stiffness.26,39 Consequently, activation of vascular MR produces target organ damage, HTN, coronary vascular dysfunction, cardiac fibrosis, alterations in renal hemodynamics, and kidney damage. Many of the effects of MR activation are not genomic.26 There is intimate interaction and synergy between genomic and non-genomic effects of aldosterone such that non-genomic effects may provide support to genomic actions.
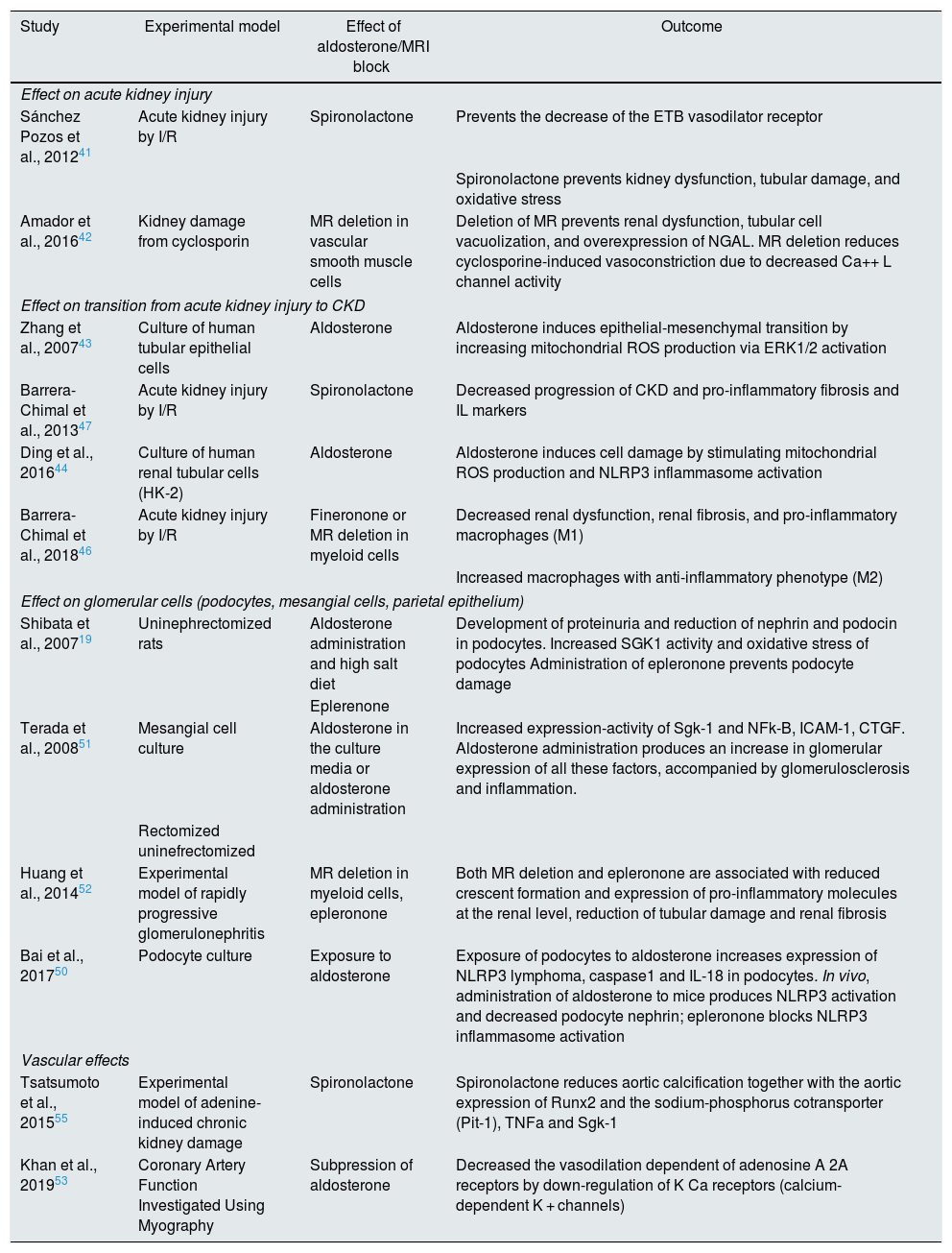
Experimental data on the effects of aldosterone and MR block in models of kidney damageThere are many studies on the effect of aldosterone and the pharmacological blockade or deletion of MR in animal models of kidney damage and in vitro investigations in various types of kidney cells (Table 2). We highlight the most relevant on the different renal structures and in the systemic vasculature.
Some experimental data on renal effects of aldosterone and mineralocorticoid receptor (MR) blockade/deletion.
| Study | Experimental model | Effect of aldosterone/MRI block | Outcome |
|---|---|---|---|
| Effect on acute kidney injury | |||
| Sánchez Pozos et al., 201241 | Acute kidney injury by I/R | Spironolactone | Prevents the decrease of the ETB vasodilator receptor |
| Spironolactone prevents kidney dysfunction, tubular damage, and oxidative stress | |||
| Amador et al., 201642 | Kidney damage from cyclosporin | MR deletion in vascular smooth muscle cells | Deletion of MR prevents renal dysfunction, tubular cell vacuolization, and overexpression of NGAL. MR deletion reduces cyclosporine-induced vasoconstriction due to decreased Ca++ L channel activity |
| Effect on transition from acute kidney injury to CKD | |||
| Zhang et al., 200743 | Culture of human tubular epithelial cells | Aldosterone | Aldosterone induces epithelial-mesenchymal transition by increasing mitochondrial ROS production via ERK1/2 activation |
| Barrera-Chimal et al., 201347 | Acute kidney injury by I/R | Spironolactone | Decreased progression of CKD and pro-inflammatory fibrosis and IL markers |
| Ding et al., 201644 | Culture of human renal tubular cells (HK-2) | Aldosterone | Aldosterone induces cell damage by stimulating mitochondrial ROS production and NLRP3 inflammasome activation |
| Barrera-Chimal et al., 201846 | Acute kidney injury by I/R | Fineronone or MR deletion in myeloid cells | Decreased renal dysfunction, renal fibrosis, and pro-inflammatory macrophages (M1) |
| Increased macrophages with anti-inflammatory phenotype (M2) | |||
| Effect on glomerular cells (podocytes, mesangial cells, parietal epithelium) | |||
| Shibata et al., 200719 | Uninephrectomized rats | Aldosterone administration and high salt diet | Development of proteinuria and reduction of nephrin and podocin in podocytes. Increased SGK1 activity and oxidative stress of podocytes Administration of epleronone prevents podocyte damage |
| Eplerenone | |||
| Terada et al., 200851 | Mesangial cell culture | Aldosterone in the culture media or aldosterone administration | Increased expression-activity of Sgk-1 and NFk-B, ICAM-1, CTGF. Aldosterone administration produces an increase in glomerular expression of all these factors, accompanied by glomerulosclerosis and inflammation. |
| Rectomized uninefrectomized | |||
| Huang et al., 201452 | Experimental model of rapidly progressive glomerulonephritis | MR deletion in myeloid cells, epleronone | Both MR deletion and epleronone are associated with reduced crescent formation and expression of pro-inflammatory molecules at the renal level, reduction of tubular damage and renal fibrosis |
| Bai et al., 201750 | Podocyte culture | Exposure to aldosterone | Exposure of podocytes to aldosterone increases expression of NLRP3 lymphoma, caspase1 and IL-18 in podocytes. In vivo, administration of aldosterone to mice produces NLRP3 activation and decreased podocyte nephrin; epleronone blocks NLRP3 inflammasome activation |
| Vascular effects | |||
| Tsatsumoto et al., 201555 | Experimental model of adenine-induced chronic kidney damage | Spironolactone | Spironolactone reduces aortic calcification together with the aortic expression of Runx2 and the sodium-phosphorus cotransporter (Pit-1), TNFa and Sgk-1 |
| Khan et al., 201953 | Coronary Artery Function Investigated Using Myography | Subpression of aldosterone | Decreased the vasodilation dependent of adenosine A 2A receptors by down-regulation of K Ca receptors (calcium-dependent K + channels) |
CTGF; connective tissue growth factor; CKD: chronic kidney disease; ETB: endothelin B; ERK1/2: extracellular signal-regulated kinases; HK-2: human kidney 2; I/R: ischemia/reperfusion; ICAM-1: intercellular adhesion molecule-1; IL: interleukins; NFk-B: nuclear factor kappa-light chain enhancer of activated B cells; NGAL: neutrophil gelatinase-associated lipocalin; NLRP3: nucleotide-binding domain and leucine-rich repeat containing PYD-3; MR: mineralocorticoid receptor; ROS: reactive oxygen species; Runx2: runt-related transcription factor 2; Sgk-1: serum and glucocorticoid regulated kinase 1; TNFalpha: tumor necrosis factor alpha.
Aldosterone causes renal vasoconstriction by increasing Rho kinase activity, endothelin, and the AT1 receptors of AII. In animal models of renal damage induced by ischemia-reperfusion, the administration of spironolactone prior to or within 6 h of the onset of ischemia attenuates renal hypoperfusion, reduces oxidative stress and prevents structural damage (tubular necrosis) and decreases tubular damage markers such as kidney injury molecule (KIM-1) and heat shock protein (HSP72).41 In the model of renal damage induced by cyclosporine, in which there is vasoconstriction of the afferent arteriole, the MR of vascular smooth muscle cells also participates by modulating the phosphorylation of their contractile proteins and the activity of their calcium L channels. Deletion of the MR of vascular smooth muscle cell prevents histological damage, vacuolization of the proximal tubular cells and overexpression of - neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage.42
Effect on the renal interstitium. Production of interstitial fibrosisThere are several mechanisms involved in the genesis of renal interstitial fibrosis and chronic kidney damage: transformation of tubular cells into mesenchymal cells, inflammation, and inadequate tissue repair after acute kidney injury (AKI). All of these have been shown to be involved in the renal profibrotic effects of aldosterone. Human proximal tubular cells exposed to aldosterone undergo epithelial-mesenchymal transformation by activation of extracellular regulated kinases (ERK1 and 2) secondary to the generation of mitochondrial ROS.43 This increase in mitochondrial ROS is also responsible for the activation of the inflammasome nucleotide-binding domain and leucine-rich repeat containing PYD-3 (NLRP3), which is as associated with an increase in fibrogenic interleukins IL-1b and IL-18 that is observed when proximal tubular cells HK-2 are cultured in aldosterone medium. Both the mesenchymal transition and the activation of the inflammasome are blocked by aldosterone antagonists.43,44
There is evidence that AKI can be a factor in the development of interstitial fibrosis and CKD.45 This AKI-interstitial fibrosis interconnection is mediated, in part, by the early appearance of inflammatory cells that play an important role in defective repair after AKI. In the model of AKI due to ischemia-reperfusion, finerenone, an MR antagonist, decreases interstitial fibrosis by activating the signals mediated by the IL-4 receptor, increasing anti-inflammatory M2 macrophages and decreasing the pro-inflammatory macrophage phenotype.46 In animal models of ischemia induced AKI, spironolactone administration, before or after ischemia, prevents the development of CKD by preventing the activation of inflammatory and profibrotic processes.47
Effect on glomerular cellsAldosterone participates in the alterations of various glomerular cells that possess MR and are able to produce functional and structural alterations of the glomerulus.
The podocyte is a fundamental component of the glomerular filtration barrier. Alterations in the function and structure of the podocyte constitute the pathophysiological basis of many glomerular diseases including diabetic nephropathy.48 The podocyte participates in aldosterone-induced glomerular damage. In uninephrectomized rat fed a high-salt diet, the administration of aldosterone induces proteinuria, structural alterations of the podocyte and a decrease in the gene expression of the podocyte proteins nephrin and podocin. Podocyte damage is associated with increased oxidative stress and SGK1 activation. Epleronone treatment prevents podocyte damage.49 In podocyte culture, aldosterone stimulates oxidative stress and the expression of SGK1, which is inhibited by epleronone and bysuperoxide dismutase, suggesting that aldosterone affects podocyte function through oxidative stress and SGK1.49
The inflammasome NLRP3 is also involved in aldosterone-induced podocyte damage. The inflammasome activates procaspase 1 which in turn induce activation of IL-1b and IL-18. In podocytes incubated with aldosterone, it is observed an increase in the expression of NLRP3 which increases the expression of caspase 1 and the concentration of IL-1b and IL-18). The increase in oxidative stress participates in the activation of NLRP3, since it is attenuated if N-acetylcysteine is added to the medium.50 Similar results are observed in in vivo experiments. The administration of aldosterone to experimental animals also induces an increase in the expression of NLPR3 in podocytes and also produces damage of the podocyte with loss of nephrin and podocin, alterations that are not observed in animals with a deletion of the NLRP3 gene.50
Aldosterone contributes to the generation of glomerular inflammation and fibrosis, two fundamental processes involved in glomerular sclerosis. Both in vivo and in vitro, aldosterone stimulates the expression and activity of SGK1 in the mesangium, which is involved in the transcription of connective tissue growth factor (CTGF) and intercellular adhesion molecule (ICAM-1), which are related to the fibrosis and glomerular inflammation, respectively. These phenomena are attenuated by the administration of epleronone.51
Aldosterone also contributes to the glomerular inflammation seen in experimental glomerulonephritis (GN). In animal models of rapidly progressive GN, deletion of MR in myeloid cells, as well as therapy with RM antagonists reduced the early neutrophil infiltration of the glomerulus and later on, the glomerular recruitment of macrophage with the corresponding decreased expression of proinflammatory cytokines, diminution of crescents and better preservation of renal function.52
Systemic vascular effectsAldosterone exerts systemic vascular effects that may be relevant in CV morbidity and mortality of CKD patients. Aldosterone increases oxidative stress, promotes phenotypic changes in vascular smooth muscle cells and fibrosis of the vascular wall.39
The infusion of aldosterone to experimental animals worsens endothelial function and vasodilation mediated by adenosine by reducing the A2a receptor dependent of calcium-activated K channels.53 This is relevant because the microvascular dysfunction evidenced by decrease in coronary reserve mediated by A2a predicts cardiovascular mortality in CKD patients on dialysis.54
Vascular calcifications are very frequent in the different stages of CKD, these calcifications have hemodynamic consequences and predict CV events and mortality. Important to note that in adenine - induced CKD animal model of vascular calcification, spironolactone administration attenuates vascular calcification by suppression of osteogenic transdifferentiation of vascular smooth muscle cells.55
Effects of MR blockade in CKD. Clinical studiesMR blockade and HTNOne of the factors that promote CKD progression is hypertension. Aldosterone favors Na+ retention, increases blood volume, produces vasoconstriction, endothelial dysfunction, increases arterial stiffness, and alters the baroreflex response. MR blockade reduces blood pressure (BP).56,57 In resistant HTN, the addition of spironolactone to the therapeutic triad: diuretic-ARS blockade-calcium antagonist produced a greater reduction of BP than the addition of doxazosin or beta-blocker.58
Resistant HTN is common in CKD. Even in CKD with well controlled BP, administration of spironolactone slows pulse wave velocity, a marker of arterial stiffness and LVH.59 The decrease in arterial stiffness benefits the heart because it reduces the central systolic BP and also favors the kidney. A rigid aorta that is not capable to mitigate the pulsatile flow generated by ventricular ejection, the fluctuations of pulsatile flow will be transmitted without attenuation to the capillaries in those organs with a low vascular resistance such as the kidney. Increased arterial stiffness is associated with a decrease in GFR.60
Plasma aldosterone values and renal expression of MR in CKDIn CKD there is an increase in plasma aldosterone which is inversely related to the GFR.61 Although, as in normal subjects, there is an inverse relationship between Na+ intake and aldosterone. However, in renal failure there is inadequate suppression of aldosterone.62 CKD is a state of relative hyperaldosteronism. The underlying cause of the increase in aldosterone when the GFR decreases, that persists despite the blockade with ACEi or ARA2, is not clear.63 There is a direct relationship between PTH/FGF23 and aldosterone, and an inverse relationship between klotho and aldosterone.64 A decrease in klotho values can increase aldosterone synthesis.15 The existence of imperceptibly elevated K+ values could also contribute to aldosterone elevation. In another sense, the association between elevated aldosterone values and a decrease in GFR could be due to the renal damage induced by aldosterone.
In the proteinuric nephropathies it has been also demonstrated increased renal expression of the MR which correlates with the presence of inflammatory markers.65
All this, together with the experimental data, reaffirms the association between CKD and aldosterone, and raises a probable nephroprotective effect of a reduction of aldosterone values or a blockade of its effects.
Aldosterone escape with drugs that block RASThe most important secretagogue of aldosterone is AII. The use and optimization of doses of drugs that block the generation or action of AII is associated with a decrease in the development and progression of CKD and death.66,67 The dual blockade of the RAS (ACEi + ARA2) reduces proteinuria compared to monotherapy, but iy is associated with a higher risk of hyperkalemia.68 In all these studies, despite therapy with ACEI, ARA2 or their combination, it persists a high residual risk of CKD progression and death.
This residual risk may be in part due to the so-called aldosterone “escape”: elevation of plasma aldosterone levels, after an initial decrease, when an RAS blocker is administered. In CKD, this occurs frequently, in 40–53% of cases,69 and it is associated with reappearance or the increase in proteinuria and a decrease in GFR.70,71 The cause of the aldosterone escape is unknown. An increase in serum K+ and ACTH, a decrease in atrial natriuretic peptide,72 or a lack of inhibition of the receptor AT1 of the A II receptor bound to ®-arrestin1 may contribute to escape.73 The involvement of aldosterone in this phenomenon is reinforced by the finding that the administration of spironolactone reduces proteinuria after its relapse.70
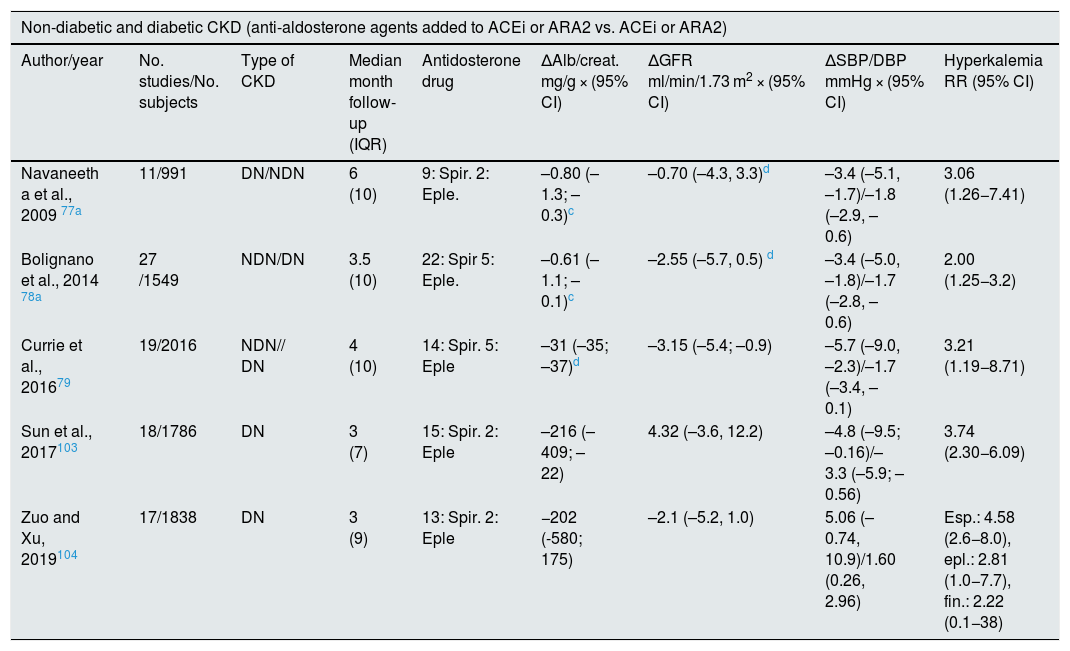
Effect of MR blockade on proteinuria and GFR in CKDThe first study on the effect of MR blockade in proteinuric nephropathy included 8 patients. The addition of spironolactone to ACEi achieved a 45% reduction in proteinuria, without changes in BP.74 In another study with 165 patients with non-diabetic proteinuric nephropathy treated with ACEI or ARA2, the administration of spironolactone 25 mg/day for one year reduced proteinuria by 50%. In the group receiving spironolactone it was observed that the GFR initially decreased with subsequent stabilization, while the control group showed a progressive decrease in GFR.75 This pattern of GFR behavior mimics that found with drugs that reduce hyperfiltration, an effect that, in the case of MR blockers, probably derives from their action on glomerular hemodynamics.36–38 In another study in subjects receiving RAS blockers with residual proteinuria, addition of spironolactone therapy for 2 years produced a progressive reduction in proteinuria, to a 63%. The evolution of the GFR was better in those in which the initial decrease was greater.76 Various meta-analyzes have shown that aldosterone antagonists added to ACEI or ARA2 produce a significant decrease in proteinuria, a reduction in BP, but an increased risk of hyperkalemia (Table 3).77–79
In an analysis of a database from subjects with CKD stages 3–4, the comparative study, after adequate adjustment of variables, between patients on spironolactone (n = 693) vs those who did not receive (n = 1386) showed that patients receiving spironolactone had less progression to CKD with less dialysis requirements (hazard ratio [HR] 0.66; 95% CI, 0.51−0.84; p < 0.001) and a higher risk of hospitalization due to hyperkalemia (HR 3.17; 95% CI 2.41–4.17; p < 0.001).80
Recently, a probable nephroprotective effect of the sodium-glucose cotransporter type 2 (iSGLT2) inhibitor dapagliflozin has been observed in non-diabetic CKD. The DAPA-CKD (The effect of dapagliflozin on renal outcomes and cardiovascular mortality in patients with chronic kidney disease) clinical trial81 included 4245 subjects with CKD stages 2–4 with increased urinary albumin excretion, with a maximum tolerated dose of ACEi or ARA2. Patients were randomized to dapagliflozin 10 mg/day or placebo. The primary endpoint was a decrease of the by GFR ≥50%, advanced CKD (FG < 15 ml/min, dialysis or renal transplant) or renal and cardiovascular death. On March 30, 2020, the trial was discontinued because it was observed, ahead of schedule, the probable benefits in the dapagliflozin branch. Although the results are not published yet, it seems presumable to attribute the early suspension of the trial to the superiority of dapagliflozin in reducing events.
One of the mechanisms involved in the nephroprotective effect of iSGLT2 is the decrease in intraglomerular pressure secondary to the vasoconstriction of the afferent arteriole induced by the increased supply of Na+ to MD.82 Coadministration of ACEi or ARA2 would enhance the decrease in intraglomerular pressure, by decreasing the vasoconstriction of the efferent arteriole. Theoretically, the administration of loop diuretics, by inhibiting Na+ reabsorption in MD, could reduce the renal hemodynamic effect of iSGLT2. However, the co-administration of antialdosteronic agents, by decreasing the distal reabsorption of Na+, inhibits the connective tubule-glomerular feedback, inducing vasoconstriction of the afferent arteriole and enhancing the effect of iSGLT2 on glomerular hemodynamics.36,37,83 Thus, the combination of ACEi or ARA2, iSGLT2 and MR antagonists could enhance the nephroprotective effects. Studies are needed to explore this hypothesis.
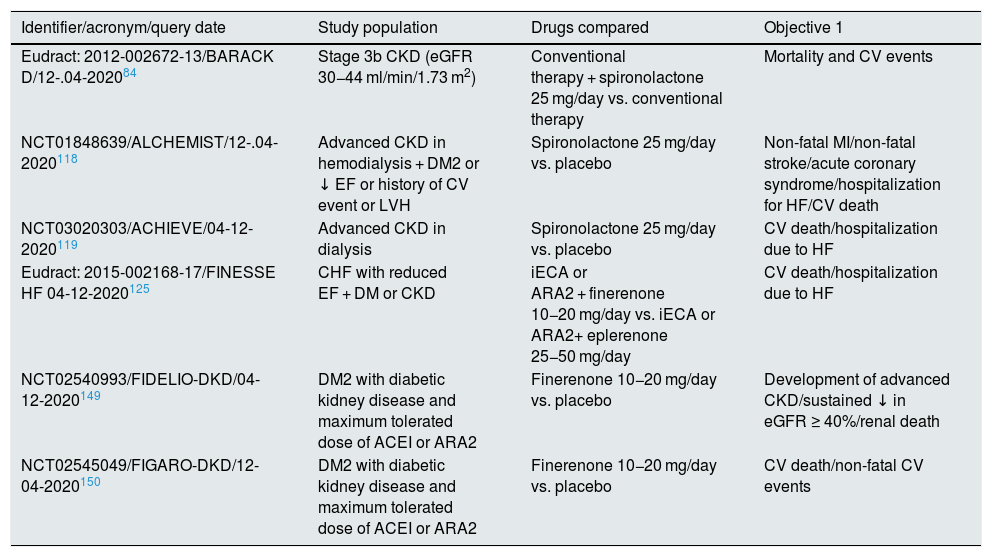
The effect of MR blockers on CV events and mortality in CKD paties is analyzed in the BARACK D84 clinical trial (Benefits of aldosterone receptor antagonism in chronic kidney disease), currently in progress, with a 3-year follow-up (Table 4).
Effect of MR blockade on proteinuria and GFR in type 2 DMMost of the referred studies included subgroups of patients with DM2. There are, however, others who have specifically looked at the effect of MR blockade on diabetic nephropathy.
The effect of ACEi, ARA2, antirenin drugs or a combination of them on the different stages of diabetic nephropathy has been evaluated.85–94 Compared with conventional antihypertensive therapy, the ARA2 reduce the risk of kidney disease progression and mortality. However still persists a high residual risk for these events.93,94
The combination ACE1-spironolactone, compared to the combination ACEi-ARA2, produces a greater decrease in proteinuria, similar effects on BP and GFR, and higher values of serum K+.95
In DM2, it is frequent to observe reduced serum levels of renin and aldosterone96,97; in this situation, it would seem reasonable to predict an attenuated response to aldosterone antagonists. However, both in animals with experimental diabetes and in subjects with DM2, the administration of spironolactone reduces alterations of podocyte and proteinuria.98,99 In an experimental model of diabetic rats with hypoaldosteronism, the administration of spironolactone prevents oxidative stress, podocyte apoptosis and proteinuria. in vitro, glucose, through MR, increases NADPH oxidase activity, ROS generation and SGK1 expression, producing podocyte damage, all of this is inhibited by spironolactone.100 The increased activity of podocyte AS and Rac1 could mediate the activation of MR by glucose, independent of the systemic aldosterone.28,29,101,102 Therefore, even in situations of low plasma aldosterone values, the administration of MR antagonists may be useful for renal protection.
Several meta-analyzes showed that in subjects with diabetic nephropathy the co-administration of an aldosterone antagonist with ACEI or ARB2, versus monotherapy with ACEI or ARB2, reduced albuminuria and BP, in addition to the increased risk of hyperkalemia, which was lower with the antagonist finerenone. of 3rd generation aldosterone antagonist103,104 (Table 3).
Meta-analysis on renal effects and mortality of antialdosterone drugs in CKD.
| Non-diabetic and diabetic CKD (anti-aldosterone agents added to ACEi or ARA2 vs. ACEi or ARA2) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Author/year | No. studies/No. subjects | Type of CKD | Median month follow-up (IQR) | Antidosterone drug | ΔAlb/creat. mg/g × (95% CI) | ΔGFR ml/min/1.73 m2 × (95% CI) | ΔSBP/DBP mmHg × (95% CI) | Hyperkalemia RR (95% CI) |
| Navaneeth a et al., 2009 77a | 11/991 | DN/NDN | 6 (10) | 9: Spir. 2: Eple. | –0.80 (–1.3; –0.3)c | –0.70 (–4.3, 3.3)d | –3.4 (–5.1, –1.7)/–1.8 (–2.9, –0.6) | 3.06 (1.26−7.41) |
| Bolignano et al., 2014 78a | 27 /1549 | NDN/DN | 3.5 (10) | 22: Spir 5: Eple. | –0.61 (–1.1; –0.1)c | –2.55 (–5.7, 0.5) d | –3.4 (–5.0, –1.8)/–1.7 (–2.8, –0.6) | 2.00 (1.25−3.2) |
| Currie et al., 201679 | 19/2016 | NDN// DN | 4 (10) | 14: Spir. 5: Eple | –31 (–35; –37)d | –3.15 (–5.4; –0.9) | –5.7 (–9.0, –2.3)/–1.7 (–3.4, –0.1) | 3.21 (1.19−8.71) |
| Sun et al., 2017103 | 18/1786 | DN | 3 (7) | 15: Spir. 2: Eple | –216 (–409; –22) | 4.32 (–3.6, 12.2) | –4.8 (–9.5; –0.16)/–3.3 (–5.9; –0.56) | 3.74 (2.30−6.09) |
| Zuo and Xu, 2019104 | 17/1838 | DN | 3 (9) | 13: Spir. 2: Eple | −202 (-580; 175) | –2.1 (–5.2, 1.0) | 5.06 (–0.74, 10.9)/1.60 (0.26, 2.96) | Esp.: 4.58 (2.6−8.0), epl.: 2.81 (1.0−7.7), fin.: 2.22 (0.1−38) |
| Advanced CKD on dialysis therapy (antialdosteronic drugs vs. placebo or conventional therapy) | |||||||
|---|---|---|---|---|---|---|---|
| Author/year | No. studies/no. subjects | Median month follow-up (IQR) | Antidosteronic | CV mortality RR (95% CI) | All-cause mortality RR (95% CI) | Hyperkalemia RR (95% CI) | RR gynecomastia (95% CI) |
| Li et al., 2019117 | 10/1172 | 6 (18) | 9: Spir. 1: Eple. | 0.42 (0.26−0.65) | 0.46 (0.32−0.66) | 1.70 (1.0−2.9) | 8.0 (2.4−26) |
| Quach et al., 2016116 | 9/829 | 6 (20) | 8: Spir. 1: Eple | 0.34 (0.15−0.75) | 0.40 (0.23−0.69) | 3.05 (1.2−7.7) | 5.6 (1.3−24)b |
Alb/creat: urine albumin/creatinine; ARA2: angiotensin II receptor antagonist; IQR: interquartile range; Eple: eplerenone; CKD: chronic kidney disease; Spir: spironolactone; ACEi: angiotensin converting enzyme inhibitor; DN: diabetic nephropathy; NDN: non-diabetic nephropathy; SBP: systolic blood pressure; DBP: diastolic blood pressure; RR (CI): relative risk (confidence interval).
Phase III clinical trials on mineralocorticoid receptor blockers in CKD, currently in progress.
| Identifier/acronym/query date | Study population | Drugs compared | Objective 1 |
|---|---|---|---|
| Eudract: 2012-002672-13/BARACK D/12-.04-202084 | Stage 3b CKD (eGFR 30−44 ml/min/1.73 m2) | Conventional therapy + spironolactone 25 mg/day vs. conventional therapy | Mortality and CV events |
| NCT01848639/ALCHEMIST/12-.04-2020118 | Advanced CKD in hemodialysis + DM2 or ↓ EF or history of CV event or LVH | Spironolactone 25 mg/day vs. placebo | Non-fatal MI/non-fatal stroke/acute coronary syndrome/hospitalization for HF/CV death |
| NCT03020303/ACHIEVE/04-12-2020119 | Advanced CKD in dialysis | Spironolactone 25 mg/day vs. placebo | CV death/hospitalization due to HF |
| Eudract: 2015-002168-17/FINESSE HF 04-12-2020125 | CHF with reduced EF + DM or CKD | iECA or ARA2 + finerenone 10−20 mg/day vs. iECA or ARA2+ eplerenone 25−50 mg/day | CV death/hospitalization due to HF |
| NCT02540993/FIDELIO-DKD/04-12-2020149 | DM2 with diabetic kidney disease and maximum tolerated dose of ACEI or ARA2 | Finerenone 10−20 mg/day vs. placebo | Development of advanced CKD/sustained ↓ in eGFR ≥ 40%/renal death |
| NCT02545049/FIGARO-DKD/12-04-2020150 | DM2 with diabetic kidney disease and maximum tolerated dose of ACEI or ARA2 | Finerenone 10−20 mg/day vs. placebo | CV death/non-fatal CV events |
ARA2: angiotensin II receptor antagonists; CV: cardiovascular; DM: diabetes mellitus; DM2: type 2 diabetes mellitus; CKD: chronic kidney disease; EF: ejection fraction; eGFR: estimated glomerular filtration rate; LVH: left ventricular hypertrophy; CHF: congestive heart failure; HF: heart failure; iECA: angiotensin converting enzyme inhibitors; MI: myocardial infarction.
Although the cited studies have some limitations, it can be concluded that in mild-to-moderate diabetic and non-diabetic CKD (stages 2–3) with proteinuria, the addition of aldosterone antagonists to ACEI or ARB2, compared with monotherapy with ACEI or ARB2, reduces proteinuria and BP, and increases the risk of hyperkalemia.
In patients with DM2, prevention of the development of nephropathy is very important. Adequate metabolic control, BP values, weight, ACEi or ARA2 and iSGLT2 are effective in reducing the appearance of albuminuria in patients with DM2.105–107 In the PRIORITY clinical trial (Proteomic prediction and renin angiotensin aldosterone system inhibition prevention of early diabetic nephropathy in type 2 diabetic patients with normoalbuminuria),108 1175 subjects with DM2 and normoalbuminuria were classified, using a urine proteomic biomarker pan (CKD273), at high and low risk of developing albuminuria. 216 subjects classified as high risk were randomized to spironolactone more conventional therapy 25 mg/day and more conventional therapy placebo. The follow-up was 2.57 years and the primary endpoint was the development of albuminuria. The results were presented at the 55th meeting of the European Association for the Study of Diabetes (EASD), held in Barcelona in September 2019. No significant reduction in the appearance of albuminuria was observed, although there was some suggestion of benefit spironolactone from the last year. The serum K+ rate > 5.5 mEq/l was higher in the spironolactone group.
There is enough evidence about the renoprotective effect of iSGLT2 in DM2109–111; given in combination with ACEi or ARA2, produces a decrease the development and progression of nephropathy in DM2. There are pathophysiological considerations, even with more argumentative force than those discussed in CKD, which raise the possibility that the triple combination of aldosterone antagonists, ACEIs or ARA2 and iSGLT2 offers greater protection in diabetic nephropathy.33,34,82,83
MR blockade in CKD patients on dialysis therapyCKD patients on HD frequently present vascular calcifications. In a small study of 5 patients on HD the administration of spironolactone 50 mg/day for 3 years produced a significant decrease in aortic calcifications.112
In CKD patients on peritoneal dialysis (PD), biopsy of the peritoneum showed that the administration of 25 mg/day of spironolactone was associated with a reduction in the amount of type IV collagen and CD20 lymphocytes in the peritoneum.113 A lesser degree of fibrosis which improves peritoneal function in these patients.
Apart from these facts, the use of aldosterone antagonists in CKD patients treated with dialysis is relevant since it has been demonstrate an association between plasma aldosterone levels and mortality, including sudden death.114
One of the largest clinical trials is the DOHAS (Dialysis outcomes heart failure aldactone study),115 in which 309 subjects on HD were randomized to receive spironolactone 25 mg/day or conventional therapy during 3 years. It was observed that in the spironolactone group there was a significant reduction in mortality or admissions for CV events 95%, from; p (HR 0.40; 95% CI, 0.20–0.80; p = 0.017).A 1.9% of the patients treated with spironolactone had severe hyperkalemia and 10.2% had gynecomastia or mastodynia.
Similar results were obtained in 2 metaanalysis that included HD and PD patients in which the effect of aldosterone antagonists was compared with placebo or conventional therapy116,117 (Table 3).
The great reduction in CV events and total mortality achieved by aldosterone antagonists in dialysis CKD patients on dialysis is prominent when compared to the observed in CKD subjects who are not on dialysis. However, the limitations of the included studies must be taken into consideration (open design, small number of patients, short follow-up, few events of CV mortality, as well as high relative risk variability demonstrated by sensitivity analysis). All this make us think that, currently, there is no definitive evidence of a benefit of MR antagonists in dialysis patients. There are two randomized trials, currently underway, may provide more certainty: one of them, ALCHEMIST (Aldosterone antagonist chronic hemodyalisis interventional survival trial), includes 825 high-risk vascular subjects with kidney failure on HD. Its duration will be 2 years and the primary objective is to analyze the effect of spironolactone 25 mg/day versus placebo on CV death and events.118 The other trial, ACHIEVE (Aldosterone blockade for health improvement evaluation in end-stage renal disease), tries to determine whether spironolactone 25 mg/day, as compared with placebo, reduces mortality and hospitalization due to heart failure (HF) in 2750 patients with CKD on dialysis treatment119 (Table 4).
MR antagonists in HF with CKDThe benefits of MR antagonists in patients with heart failure with reduced eyection fraction (EF) are well established.7–9 Even in HF with preserved EF, MR antagonists can reduce admissions for heart failure.120 More than 50% of patients with heart failure have CKD and their mortality increases with the decrease in GFR.121 A large percentage of patients with heart failure and CKD do not receive MR blocker therapy because of the concern of hyperkalemia.122 In the RALES trial, the relative risk of total mortality and hospitalization for heart failure in subjects treated with spironolactone was similar in those with and without a decrease in GFR (0.68, 95% CI, 056−0.84 and 0.64; 95% CI, 0.52−0.72 vs 0.71, 95% CI, 0.57−0.90 and 0.67, 95% CI, 0.56−0,81, respectively). The reduction of absolute risk for mortality and admission for heart failure was greater in the presence of renal failure. As expected, the aldosterone antagonists produced hyperkalemia more frequently if the GFR was reduced (25.6% vs 15.4%)123.
Non-steroidal MR blockers with a better safety profile (less hyperkalemia) and new intestinal K+ linkers may facilitate the use of aldosterone antagonists in CKD patients with heart failure.121,124
The ongoing FINESSE clinical trial compares the effect of finerenone versus eplerenone on CV mortality and admission for heart failure in subjects with chronic heart failure with reduced EF and DM-2 or CKD125 (Table 4).
MR antagonists and hyperkalemiaIn contrast to the consistent evidence of the CV and mortality benefits of aldosterone antagonists in cardiology7–9, there are few specific studies of the effect of these drugs on these events in nephrology. It is possible that one of the hyperkalemia, especially in situations of renal failure, has been a constrain for the frequent use of these drugs in CKD.
As observed in clinical trials and in real life studies, the combination of aldosterone antagonists with ACEI or ARA2 increases the risk of hyperkalemia.104,116,126
This fact is important since in conditions frequently associated with abnormal regulation of K, such as heart failure, DM and CKD, there is a U shape relationship between serum K+ and mortality.127
In subjects with less capacity to excrete K+ in the urine due to a reduction in the diuresis, aldosterone antagonists also increase serum K+. Compared with subjects with normal renal function, patients with severe CKD have an increased expression of K+ channels (BK channels) in colon enterocytes that increases their capacity for intestinal elimination of K+.128 Aldosterone stimulates colonic K+ secretion through BK channels, secretion that is inhibited by spironolactone.129,130
The association of aldosterone antagonists with ACEI or ARA2 contributes to hyperkalemia, but in these patients there may also concur other causes of elevated serum K+.131
Given the occurrence of hyperkalemia in subjects who receive RAAS blockers for processes in which they have shown benefit, frequently the dose has to be reduced or even suppressed. This fact may condition an increase in CV and renal events, and mortality.132 The use of the new oral K+ binders as patiromer and zirconium cyclosilicate is likely to improve tolerability and minimize discontinuation of RAAS133–135 blockers. On the other hand, the new non-steroidal aldosterone antagonists induce less hyperkalemia than the classic antagonists.136
Nonsteroidal aldosterone antagonistsMost studies with aldosterone antagonists have used the classic steroid antagonists: spironolactone, which has low specificity of action on MR, and eplerenone, which is more selective.
Nonsteroidal aldosterone antagonists have a different chemical structure from that of steroids, which determines differences in physicochemical properties, pharmacological actions, tissue penetration and distribution, the mode of binding to the ligand and the ability to bind to co-regulators, among others.137 All this may condition special effects on organ protection and attenuation of adverse effects.
To date, 7 nonsteroidal aldosterone antagonists has been tested in clinical trials in different phases, some of them being discontinued for various reasons. Others have completed phase 1 trials or are recruiting patients for phase 2.138–141
The most advanced nonsteroidal aldosterone antagonists are: apararenone (MT3995, Mitsubishi Tanabe Pharma Corporation, Osaka, Japan), exaserenone (CS3150, Exelisis, California, USA, and Daiichi Sankio, Tokyo, Japan) and finerenone.
Aphase 2 clinical trial with Apararerone has completed in subjects with diabetic nephropathy.142 Esxerenone, whose maximum 50% inhibitory concentration (IC50) of MR transcriptional activation is 9.4 nM, is a more potent, more selective and specific inhibitor than spironolactone and epleronone. In a randomized clinical trial, Esxerenone has been shown to be effective and well tolerated in patients with HTN.143
The finerenona has an IC50 of 18 nM (much lower than spironolactone and eplerenone and therefore more powerful). It is much more selective for MR than spironolactone and epleronone and, unlike those predominantly distributed in the kidney, finerenone is equally distributed in the kidney and heart.136
Experimental studies show that finerenone produces renal and vascular protection by reducing oxidative stress and attenuating endothelial dysfunction, among other effects.144,145
In the clinical trial ARTS (The mineralocorticoid receptor antagonist tolerability study), there were 782 included with heart failure and reduced EF and CKD stages 2−3. Fineronona 5−10 mg/day, compared with spironolactone 25−50 mg/day, produced the same effect on the reduction of NT-pro-BNP and albuminuria, less deterioration of renal function and less elevation of serum K+ with a lower incidence of hyperkalemia (5% vs. 13%) 146.
In a similar patient population, different doses of finerenone, compared with epleronone 25−50 mg/day, produced a similar reduction in NT-pro-BNP and a lower increase in serum K+; and in doses of 20 mg/day, finerenone produces a greater reduction in CV events and mortality than epleronone. However, as it was a short study, these variables were considered exploratory.147
In the test ARTS-DN (Minerlocorticoid receptor antagonist tolerability study- diabetic nephropathy), 823 patients with diabetic nephropathy treated blockers of the Renin Angiotensin system (ACEi or ARA2) were randomized to different doses of finerenona or placebo. The main objective was the modification of proteinuria. Fineronone induced a significant dose-dependent reduction in proteinuria (50% reduction in proteinuria in 40% of patients with 20 mg/day), with a slight non-significant decrease in GFR, reversible after stopping the drug, and a decrease in BP. It was observed a low frequency of hyperkalemia (K+ > 5.6 mEq/l) (1.6% of the group treated with 20 mg/day of finerenone vs. 1.5% of the placebo group).148
These are short-term studies that analyze the effect of nonsteroidal aldosterone antagonists on intermediate variables. In patients with heart failure and reduced EF, the Classic MR antagonists have achieved benefits relative to CV events and mortality despite an increase in hyperkalemia. Activation of MR produces harmful cardiorenal effects by various mechanisms that have been discussed in this review. It is possible that nonsteroidal aldosterone antagonists, with a theoretical greater cardiorenal protective capacity and with less risk of hyperkalemia, more benefits will be obtained in important CV and renal variables.
The definitive answer will be provided by 2 clinical trials with finerenone. The FIDELIO-DKD (Finerenone in reducing kidney failure and disease progression in diabetic kidney disease) trial investigates the efficacy of finerenone compared to placebo in reducing major renal and CV events in subjects with DM-2 and CKD treated with ACEI or ARA2. The primary endpoint is the time elapsed until the onset of advanced kidney disease, decreased eGFR sustained ≥40% or renal death149. There were 5734 patients randomized, ending the follow-up in 2020. The study has been published recently. Finerenone reduced the risk of the composite cardiovascular outcome compared with placebo (hazard ratio, 0.86 [95% CI, 0.75−0.99]; p = 0.034), with no significant interaction between patients with and without CVD. The incidence of treatment-emergent adverse events was similar in both arms.
The FIGARO- DKD (Finerenone in reducing CV mortality and morbidity in diabetic kidney disease) investigates, in subjects with DM2 and CKD, the efficacy and safety of finerenone compared with placebo, to reduce clinically relevant CV and renal events.150 The primary endpoint is the composite of CV death, non-fatal myocardial infarction, non-fatal stroke, and hospitalization for heart failure. 7437 patients were recruited, currently under follow-up, waiting for results in 2021.
Key concepts- 1
In addition to the classic sites, such as distal renal and connecting tubule, and colon, there are MR in other locations, such as podocyte, mesangium, macula densa, endothelium, vascular smooth muscle cell, myocardiocyte, adipocyte, macrophage, among others. In certain situations, MR can be activated by mechanisms independent of aldosterone.
- 2
Activation of MR by aldosterone increases distal renal tubular Na+ reabsorption and K+ secretion. In addition to systemic hemodynamic effects (increased BP and arterial stiffness), aldosterone modifies intraglomerular hemodynamics by modulating the tubulo-glomerular and connecting tubule-glomerulus feedbacks.
- 3
Activation of vascular MRI (endothelium, smooth muscle fiber) produces increased oxidative stress, endothelial dysfunction, fibrosis, vascular remodeling, and increased arterial stiffness.
- 4
In experimental models, aldosterone and MR activation produce kidney damage by various mechanisms (Fig. 2).
- 5
In CKD with decreased GFR and proteinuria, there is an increase in plasma aldosterone values and in renal expression of MR.
- 6
A high percentage of subjects with CKD treated with ACEi or ARA2 present aldosterone leakage, which is associated with a reappearance or increase in proteinuria and a greater decrease in GFR.
- 7
Many short-follow-up studies have shown that in diabetic and non-diabetic CKD patient with proteinuria, classic aldosterone antagonists (spironolactone, eplerenone) reduce proteinuria and increase the risk of hyperkalemia. There are several clinical trials currently underway that attempt to study the effect of spironolactone administration on mortality and CV events in subjects with CKD stages 3b and 5d.
- 8
ACEi and ARA2, and the association of iSGLT2, have been shown to improve nephroprotection in diabetic and non-diabetic nephropathy. There are pathophysiological considerations that raise the possibility that the combination of ACEi or ARA2, iSGLT2, and MR blockers may enhance nephroprotection.
- 9
Compared with steroidal MR blockers, the more modern non-steroidal MR blockers have higher potency and selectivity, and produce less elevation of serum K+. The underway clinical trials (FIGARO-DCK and FIDELIO-DCK just completed) will determine whether in subjects with DM2 and nephropathy and with a maximum tolerated dose of ACEi or ARA2, the non-steroidal MR blocker, finerenone, 10−20 mg/day, reduces mortality, CV events and the progression of kidney disease.
.")
Dr. Pablo Gómez-Fernández has participated as principal investigator in the following Bayer-sponsored trials.
- -
Mineralocorticoid rec eptor antagonist tolerability study-diabetic nephropathy (ARTS-DN; ClinicalTrials.gov identifier NCT01874431).
- -
Finerenone in reducing kidney failure and disease progression in diabetic kidney disease (FIDELIO-DKD, ClinicalTrials.gov identifier: NCT02540993).
- -
Finerenone in reducing CV mortality and morbidity in diabetic kidney disease (FIGARO-DKD; ClinicalTrials.gov identifier NCT02545049).
Please cite this article as: Erraez S, López-Mesa M, Gómez-Fernández P. Bloqueantes del receptor mineralcorticoide en la enfermedad renal crónica. Nefrologia. 2021;41:258–275.



