




Chronic kidney disease (CKD) is a pathology with a high worldwide incidence and an upward trend affecting the elderly. When CKD is very advanced, the use of renal replacement therapies is required to prolong its life (dialysis or kidney transplantation). Although dialysis improves many complications of CKD, the disease does not reverse completely. These patients present an increase in oxidative stress, chronic inflammation and the release of extracellular vesicles (EVs), which cause endothelial damage and the development of different cardiovascular diseases (CVD). CKD patients develop premature diseases associated with advanced age, such as CVD. EVs play an essential role in developing CVD in patients with CKD since their number increases in plasma and their content is modified. The EVs of patients with CKD cause endothelial dysfunction, senescence and vascular calcification. In addition, miRNAs free or transported in EVs together with other components carried in these EVs promote endothelial dysfunction, thrombotic and vascular calcification in CKD, among other effects. This review describes the classic factors and focuses on the role of new mechanisms involved in the development of CVD associated with CKD, emphasizing the role of EVs in the development of cardiovascular pathologies in the context of CKD. Moreover, the review summarized the EVs’ role as diagnostic and therapeutic tools, acting on EV release or content to avoid the development of CVD in CKD patients.
La enfermedad renal crónica (ERC) tiene una alta incidencia mundial y una tendencia ascendente que afecta principalmente a personas de edad avanzada. Cuando la ERC está muy avanzada se requiere el uso de terapias renales sustitutivas para prolongar la vida (diálisis o trasplante renal) y, pese a que la diálisis mejora muchas complicaciones de la ERC, la enfermedad no revierte de manera completa. Estos pacientes presentan un aumento del estrés oxidativo, inflamación crónica y aumento de la liberación de vesículas extracelulares (VE), que provocan daño endotelial y el desarrollo de distintas enfermedades cardiovasculares (ECV). De hecho, los pacientes con ERC desarrollan de forma prematura enfermedades asociadas a una edad avanzada, como es el caso de las ECV. Las VE desempeñan un papel muy importante en el desarrollo de ECV en pacientes con ERC, ya que su número aumenta en el plasma y su contenido se modifica. Las VE de pacientes con ERC generan disfunción endotelial, senescencia y calcificación vascular. Además, los miRNA libres o transportados en las VE junto a otros componentes vehiculados en estas VE promueven disfunción endotelial, eventos trombóticos y calcificación vascular en los pacientes con ERC, entre otros efectos. En esta revisión se describen los factores clásicos y el papel de nuevos mecanismos que intervienen en el desarrollo de la ECV asociada a la ERC, con especial hincapié en el papel de las VE en el desarrollo de enfermedades cardiovasculares en un contexto de ERC. Además, se expone el papel de las VE como herramienta diagnóstica y como diana terapéutica, actuando sobre su liberación o contenido para intentar evitar el desarrollo de ECV en enfermos renales crónicos.
Chronic kidney disease (CKD) is considered a public health problem since it affects approximately 850 million people worldwide and it is responsible for approximately 2.4 million deaths per year; in fact, in developed countries, around 11% of the population suffers from CKD.1 In Spain, around 7 million people suffer from this pathology, with a notable increase –around 4%–5% each year– in the last decade.2,3 This increase seems to be closely related to different causes, among which the aging of the population stands out. Nowadays, around 7% of the world’s population is over 65 years of age, approximately 15% in developed countries. In Spain, around 35% of the population is over 75 years of age.4 Another possible cause is the increase in the prevalence of other important risk factors such as obesity, arterial hypertension (AHT), smoking and diabetes mellitus (DM). This last disease is the most frequent cause of CKD, accounting for 26.54% of new cases of CKD5 in Spain in 2020 (Fig. 1) and mortality due to CKD worldwide is 44.7%.3

Chronic kidney disease, associated diseases and therapies. CKD is caused by diseases such as diabetes mellitus, polycystic kidney disease or arterial hypertension, among others. As a consequence of CKD, diseases related to vascular damage and CVD occur, which are mediated by different factors. The main treatment modalities in patients with CKD are shown below.
The loss of renal function in patients with CKD is progressive and silent. Symptoms of CKD often do not appear until the disease is very advanced (advanced chronic kidney disease; ACKD), even shortly before patients require renal replacement therapy (RRT).6 RRT using dialysis helps to prolong lives of many end-stage patients, although it can also complicate their clinical condition by worsening cardiovascular function.7,8 There are two dialysis modalities: hemodialysis (HD) and peritoneal dialysis (PD). Both techniques manage to eliminate part of the uremic toxins accumulated in the blood. The ideal therapy that can solve this condition is kidney transplantation, although this requires find a kidney from a compatible donor, which can take months or even years, and even then it is not a definitive solution. A significant percentage of patients are left with a certain degree of chronic kidney failure; and the average life expectancy of a kidney transplant is under 20 years9 (Fig. 1).
In patients with CKD it is common to observe different comorbidities, especially cardiovascular diseases (CVD), which are the main cause of death.7,8 The increase in mortality due to CVD occurs from the early stages of CKD10 triggered by the endothelial damage generated by CKD11 (Fig. 1). Controlling the different comorbidities and risk factors that can accelerate the decline in renal function is essential for slowing the development of CKD, as well as its early detection, which continues to be a challenge today.5
The high risk of developing CVD in uremic patients with CKD makes it necessary to identify new biomarkers for the prediction and diagnosis of cardiovascular complications and the development of new therapies for the treatment of these pathologies.12 In recent years, different elements and mechanisms have been identified that may be involved in the early development of CKD and/or CVD, among which extracellular vesicles (EV), microRNA (miRNA), or the count of certain immune populations stand out.11
The aim of this study is to review what is known of the mechanisms that determine the onset of CVD in patients with CKD. Aspects involved in the accelerated aging of patients with CKD and, more specifically, with the development of vascular damage will be analyzed, including the role of EVs as an important effector associated with the development of complex illnesses in CKD. The early identification of biomarkers involved in these processes is of great interest in order to find new therapeutic targets and to prevent and/or avoid the development of CKD and associated CVD.
The development of cardiovascular disease in patients with chronic kidney diseaseThere are numerous connections between CKD and CVD; in fact, they share risk factors such as HTN, present in most renal patients, DM and dyslipidemia,5 which suggests that the development of CVD associated with CKD has a multifactorial origin.
The loss of renal function induces metabolic and biochemical alterations. One of the most important alterations is the accumulation of uremic toxins, which, when bound to proteins, cannot be eliminated with the dialysis treatments due to their high molecular weight and will trigger CVD.13 In addition, these toxins will produce vascular damage such as fibrosis of the tunica intima, hyperplastic arteriosclerosis (related to HTN), atherosclerotic plaque formation and vascular calcification of the tunica media. These last two manifestations are associated with greater morbidity and mortality in patients with CKD.14 What is more, the accumulation of uremic toxins in patients with CKD causes endothelial dysfunction: increased vascular permeability due to loss of cell–cell junctions, increased oxidative stress, and activation of various proinflammatory and prothrombotic signaling pathways.15
Dialysis can also contribute to the development of CVD due to the use of dialysis fluids, immunosuppressants, anticoagulants among others.16 These therapies are associated with an increase in oxidative stress and systemic inflammation which, if persistent over time, alters the immune response in renal patients. Likewise, platelet activation, activation of the complement cascade and activation of the immune system occurs in this type of patient. All these factors will affect the endothelium and result in damage of the vasculature, a preliminary step that triggers multiple vascular pathologies associated with CKD.17
Endothelial damage associated with chronic kidney diseaseThe endothelium is more than just a barrier between the bloodstream and the vascular wall; it is an active component involved in numerous processes, including regulation of vascular tone, growth and migration of the underlying smooth muscle cells, and maintenance of vascular structure.18 Under physiological conditions, endothelial cells present an anti-adherent and anti-coagulant surface; but in response to damage, the molecules expressed on their surface can be modified and thus increase cell adhesion capacity. Platelets bind to the damaged surface initiating the coagulation process with the consequent development of inflammation and thrombosis, causing cardiovascular events. Hence, endothelial damage can be the initial event that triggers the appearance of different cardiovascular pathologies11 (Fig. 2).

Vascular damage. Oxidative stress and inflammation, among other factors, generate endothelial deterioration leading to: increased adhesion capacity that causes thrombosis, increased EV release and increased leukocyte extravasation, which favors the development of cardiovascular disease. Figure elaborated with Biorender.
There are different mechanisms involved in the deterioration of the endothelium; the most important in patients with CKD are described below.
Oxidative stressOxidative stress is defined as the accumulation of oxidizing molecules, mainly reactive oxygen species (ROS), either by an increase in their production or by a decrease in the antioxidant mechanisms. ROS have a high oxidative capacity and can therefore alter different molecules (proteins, lipids, nucleic acids, etc.) in the cells, causing damage.19,20 In early stages of CKD, it has been observed a correlation between high levels of oxidative stress and disease progression.21,22 Furthermore, patients with CKD accumulate uremic toxins in their bloodstream, including IS (indoxyl sulfate), MDA (malondialdehyde) and ADMA (asymmetric dimethylarginine), which worsen the oxidative stress.23
In general, oxidative stress can be produced by various causes (Fig. 3). In patients with CKD, mainly those with diabetic nephropathy, there is mitochondrial dysfunction that leads to increased ROS production. Studies in animal models and in vitro studies, have shown that in the presence of IS there is an increase in the activity of the enzyme NADH oxidase 4 (NOX4), which leads the generation of larger amounts of ROS.23

ROS also alter cellular structures and metabolic pathways. One of the markers used to measure oxidative stress are the so-called AGEs (advanced glycation end products). AGEs bind to their RAGE receptors (receptor system for AGE) and signal through the MAP kinase (MAPK) pathway. Activation of MAPKs allows internalization of the p65 subunit of NF-κB (nuclear factor kappa B) into the nucleus and consequently, increases the release of proinflammatory cytokines and enzymes, and also increase the expression of adhesion molecules.24 At the same time, activated pro-inflammatory immune cells, release oxidative compounds, thus generating a vicious cycle that exacerbates oxidative damage.25
Increased oxidative stress damages DNA. Guanine in particular can be oxidized and transformed into 8-hydroxy-2′-deoxyguanosine (8-OH-dG), which is another marker used to measure oxidative stress.26 These changes in DNA bases generates cellular damage and has been associated with multiple chronic and degenerative diseases, including CKD and carcinomas.27 In addition, there are numerous factors that gradually increase in CKD due to the deterioration of renal function, and these factors are considered to be responsible for the increase in oxidative stress (Table 1).
Oxidative stress associated to CKD.
| Parameter | Result | Patients | Sample | Reference |
|---|---|---|---|---|
| GSH | ↓ In patients with moderate or severe CRF in preD. | 31 HC83 patients with mild chronic renal failure (CRF) in pre-D,55 patients with moderate CRF in preD47 patients with severe CRF in preD18 HD patients | WB | 28 |
| GSSG | ↓ In HD compared to HC and severe CRF | |||
| GSH-Px activity | ↑ In all preD compared with HC | E | ||
| GSSG-Red activity | ↑ In all preD compared to HC↓ In HD than in patients with severe CKD in preD | |||
| GSH-Px activity | ↓ Gradually as renal failure progresses in preDCAPD↓ In HD and CAPD | Plasma | ||
| GSSG-Rd activity | ↑ In HD and CAPD patients compared to HC and patients with severe CRF inpreD | |||
| SOD activity | ↓ In HD patients | E | ||
| MDA | ↑ In patients with CKD compared to HC,but are maintained once the disease is started | 31 HC 73 patients with mild CRF53 withmoderate CRF36 with advanced CRF | Plasma | 29 |
| GSH-Px activity | ↓ According to progression of renal damage | |||
| AOPP | ↑ In patients with moderate and advanced CKD | |||
| AGE-pentoside | ↑ As renal damage progresses | |||
| CRP | ↑ In patients with CKD | 70 HC60 CKD patients | Plasma | 30 |
| Thiols | ↓ In CKD patients | |||
| Carbonyls | ↑ In CKD patients | |||
| F2-isoprotans | ↑ In CKD patients | |||
| IL-6 | ↑ In CKD patients | |||
| ROS | ↑ In CKD patients | 21 CS PBMNs 22 patients with CKD in stages 1 and 2 | PBMNs | 31 |
| NADPH oxidase activity | ↑ In CKD patients | |||
| AOPP | ↑ In HD compared to PD | 1539 HD patients556 PD patients | Serum | 32 |
| Vitamin C | ↓ In CKD and even lower in HD patients | 38 HC51 patients with CKDstages 3–550 HD | Plasma | 33 |
| Zinc | ↓ In CKD and HD compared to HC | E | ||
| SOD activity | ↓ In HD patients | |||
| XO activity | ↑ In CKD and even more in HD | |||
| MDA | ↑ In HD |
AGE: advanced glycation products; AOPP: advanced glycation protein products; CAPD: continuous ambulatory peritoneal dialysis; HC: healthy controls; PD: peritoneal dialysis; E: erythrocytes; CKD: chronic kidney disease; CRF: chronic renal failure; GSH: glutathione; GSH-Px: glutathione peroxidase; GSSG: glutathione disulfide; GSSG-Red: glutathione reductase; HD: hemodialysis; IL-6: interleukin 6; MDA: malondialdehyde; PBMN: peripheral blood mononuclear cells; CRP: C-reactive protein; preD: predialysis; ROS: reactive oxygen species; WB: whole blood; SOD: superoxide dismutase; XO: xanthine oxidase.
Hyperphosphatemia, present in many patients with CKD, also plays an important role in vascular deterioration and it is related to oxidative stress. In vitro experiments with endothelial cells showed that high phosphate concentrations generate an increase in oxidative stress and a drop in nitric oxide (NO). Under pathological conditions there is a decrease in NO synthesis as a consequence of inhibition of the phosphorylation of the endothelial nitric oxide synthase (eNOS).34 Under physiological conditions NO is released by endothelial cells to produce vascular relaxation and prevent arterial stiffness, so that a decrease in its production favors endothelial deterioration. These results have been corroborated by experiments carried out in healthy and CKD mice: diets with a high content of phosphate promote inflammation and endothelial dysfunction, in addition to a loss of permeability between endothelial cells.35
Numerous studies show how oxidative stress is directly related to proinflammatory factors.36–38 Patients on HD may show an increase of up to 30–40 times the physiological concentration of acetate after the completion of the dialysis session, acetate is the most frequent buffer used in dialysis fluids. This elevation in blood acetate is associated with an increase in oxidative stress, as well as with an increase in some proinflammatory cytokines such as interleukins 1 and 6 (IL-1 and IL-6) and with the synthesis of NO.36–38 If there is also a high concentration of magnesium in the dialysis fluid (1.25 or 2mM), the in vitro production of ROS and MDA increases, although this increase in magnesium has been seen able to prevent an increase in oxidative stress when citrate is used instead of acetate as in dialysis fluids (citrate alone or with a mixture of citrate and a small amount of acetate).16 The increase in the concentration of oxidized glutathione as a result of HD means that the dialysis technique is considered a risk factor associated with an increase in oxidative stress.39
Thus, in patients with CKD uremia triggers an increase in oxidative stress that particularly affects endothelial cells due to their direct contact with blood. In turn, this oxidative stress generates alterations that will favor the development of inflammatory processes; in such a way that the coexistence of inflammation and oxidative stress will trigger and enhance endothelial dysfunction in patients with CKD.
Inflammatory mediators and immune response in CKDInflammation plays an important role in the increased risk of cardiovascular events in patients with CKD. It has been shown that inflammatory processes are triggered by factors such as HTN, advanced age, DM, obesity, hyperuricemia, dyslipidemia, smoking, male sex, and family history of CVD.40 Similarly, both the accumulation of uremic toxins and also dialysis procedure required to remove uremic toxins promote the development of inflammation.40,41
Patients with CKD present chronic inflammation caused by uremia and dialysis techniques.42 These inflammatory processes in dialysis patients are more frequent in advanced stages of the disease. Particularly important is the continuous damage of the peritoneum produced during PD that produces the activation of genes related to adaptive immunity and the Th2 lymphocyte response.43 Regarding HD, the use of non-biocompatible membranes or the possible contamination of the dialysis fluid, among other factors,44 will trigger the activation of monocytes, which release proinflammatory cytokines. However, the presence of inflammation in pre-dialysis patients tell us that indicates monocyte activation is not the main cause of inflammation.45–47
At the onset of CKD, patients present low-intensity systemic inflammation which, when prolonged over time, leads to a deterioration in the general condition of the organism and favors the appearance of secondary pathologies such as CVD.48 It is observed a slight elevation of inflammatory markers such as C-reactive protein (CRP) and some cytokines such as IL-6 and tumor necrosis factor α (TNF-α)49; CRP can even be considered a marker of mortality, it has been observed that lower levels of CRP are associated with a lower risk of mortality in HD patients.50 Nevertheless, IL-6, IL-1 and TNF-α are directly related to the severity of CKD. IL-6 has been described as a predictive marker of atherosclerosis, since it contributes to the generation and development of atheroma plaque by various mechanisms.51 The specific signaling pathways used by IL-6 in the development of arteriosclerosis are unknown. In animal models it has been observed that upon binding IL-6 to its receptor IL6-R, the IL-6 trans signaling pathway is activated, which triggers a chronic inflammatory process and favors the formation of atheroma plaque, and therefore, the development of cardiovascular disease.52 IS can bind to the aryl hydrocarbon receptor (AhR) of monocytes to stimulate the secretion of TNF-α. Then TNF-α interacts with endothelial cells increasing the expression of the chemokine CX3CL1 (also known as fractalkine), whose receptor is CX3CR1, is abundant in CD4+CD28− T lymphocytes. This subpopulation of T lymphocytes is elevated in patients with CKD and, if stimulated through the T-cell receptor (TCR), can induce apoptosis in endothelial cells and accelerate the progression of CVD in these patients53 (Fig. 4).

Endothelial dysfunction and vascular calcification generated by IS binding to the AhR receptor on monocytes. The increase in TNF-α generated by IS generates an increase in the production of the chemokine CX3CL1 in endothelial cells. When CX3CL1 binds to its receptor on the T lymphocyte, and the T lymphocyte has its T cell receptor active, it will generate the development of endothelial cell apoptosis. AhR: aryl hydrocarbon receptor; BMP2: bone morphogenic protein 2; IS: indoxyl sulfate; MSX2: Msh homrbox protein 2; Runx2: runt-related transcription factor 2; TCR: T-cell receptor; TNF-alpha: tumor necrosis factor alpha; TNFR: TNF receptor.
TNF-α is one of the main activators of RANKL (receptor activator ligand for nuclear factor κB). Thus, TNF-α in the bone tissue of patients with CKD can activate osteoclasts, altering bone structure and increasing the risk of fractures, which explains the high frequency of fractures in patients on HD.54 Furthermore, the activation of osteoclasts prompts an increase in blood levels of calcium and phosphate which facilitates the mineralization processes of vascular cells. TNF-α potentiates this mineralization process since it allows the translocation of NF-κB, increases the release of EV (which in renal patients contain high levels of calcium) and increases the levels of bone morphogenic protein 2 (BMP-2), inducing osteogenic differentiation and calcification of vascular smooth muscle cells (VSMC).55
In patients with CKD, activated endothelial cells also express a greater amount of VEGF (vascular endothelial growth factor) which, in turn, promotes angiogenesis and vessel remodeling to counteract renal tissue hypoxia. Likewise, in patients who present proteinuria due to a decrease in glomerular filtration, it has been seen that the endothelial cells of the glomerulus increase the release of VEGF.56,57
Extracellular vesiclesThe accumulation of uremic toxins has been associated with endothelial damage resulting, among other effects, in increased release of EVs.58,59 EVs serve as a signaling system at a local and systemic level, and are involved in the function and homeostasis of the organism.60 In general, they are very diverse and can be found in all body fluids, including breast milk and urine.61 EVs are produced by most cells, including B cells, T cells, monocytes and endothelial cells. The formation of EVs is part of normal cellular function; however, EVs production increases under conditions of cellular stress, apoptosis or altered cell viability, as is the case in CKD.62
EVs can be classified according to their origin, composition and size into: A) exosomes (40–120nm), the product of exocytosis of multivesicular bodies; B) microvesicles (MV) (50–1000nm), also known as microparticles, which arise from evaginations of the plasma membrane; and, C) apoptotic bodies (1–5μm), which are formed in the final stages of apoptosis.11,40 For practical purposes, due to the difficulty in isolating them and the lack of a protocol to obtain a single type of EV, it is almost impossible to distinguish one type from the other, especially between the first two. For this reason, they are now generally referred to as EVs, without distinguishing between types.
In the kidneys, EVs have been closely linked to pathological processes such as inflammation, fibrosis, thrombosis, and immune suppression.63 MVs and exosomes may actively play pathological roles in the development and progression of CVD because they act as carriers of various miRNAs.40 In addition, they are involved in the development of vascular calcification due to the accelerated vascular aging state.64 Therefore, EVs and their content have been proposed as therapeutic targets, since they could be inhibited to reverse and/or treat the progression of kidney disease. Furthermore, EVs may be involved in various processes of tissue repair and immune modulation, as they could be directly used as therapeutic agents in regenerative medicine and the treatment of autoimmune diseases.
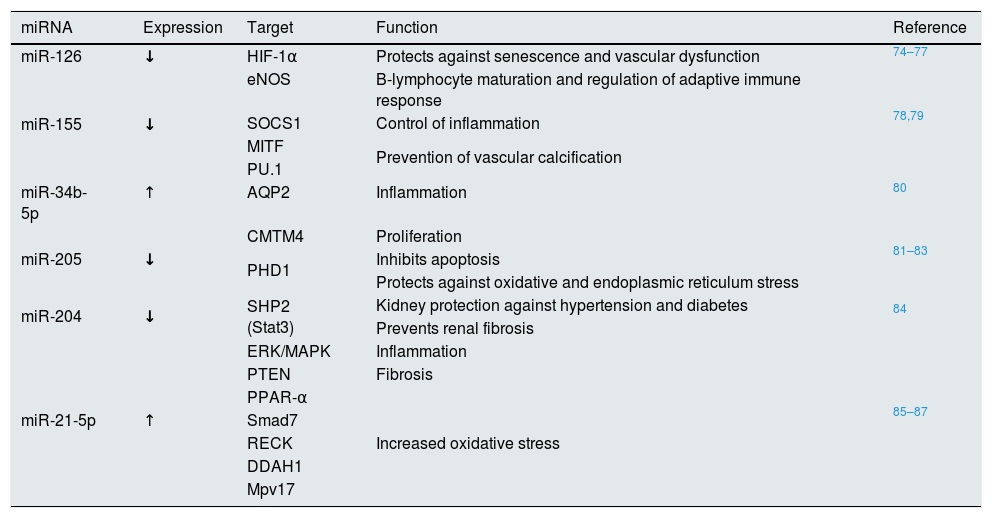
microRNAEVs transport proteins, lipids and nucleic acids, among which miRNAs stand out, although the latter can also be transported freely in the blood.65 The miRNAs are small non-coding RNAs (approximately 22 nucleotides long), whose main function is the regulation of protein expression.66 The miRNAs act in two ways: they can partially inhibit protein expression by binding to the 3′ UTR region of the transcript, where they initiate the degradation of the messenger RNA and inhibit its translation; or they can interact with the promoter, favoring the recruitment of the gene transcription machinery and, therefore, increasing protein expression.67,68 Most miRNAs are present inside the cell, although they can also be found in body fluids such as blood.69,70 To avoid their degradation by enzymes with RNase activity, it is suggested that these extracellular miRNAs are packaged within EVs, mainly exosomes and MV.69,71,72 miRNAs are highly tissue and cell-type specific, even specific to developmental stage, and they are involved in a large number of pathophysiological processes including inflammation. In the vascular environment, miRNAs act as regulatory molecules in endothelial cells, VSMCs, platelets and other blood cells.73Table 2 lists the most relevant miRNAs whose expression is being modified in CKD.
miRNAs involved in vascular pathologies whose expression is modified in patients with CKD.
| miRNA | Expression | Target | Function | Reference |
|---|---|---|---|---|
| miR-126 | ↓ | HIF-1α | Protects against senescence and vascular dysfunction | 74–77 |
| miR-155 | ↓ | eNOS | B-lymphocyte maturation and regulation of adaptive immune response | 78,79 |
| SOCS1 | Control of inflammation | |||
| MITF | Prevention of vascular calcification | |||
| PU.1 | ||||
| miR-34b-5p | ↑ | AQP2 | Inflammation | 80 |
| miR-205 | ↓ | CMTM4 | Proliferation | 81–83 |
| PHD1 | Inhibits apoptosis | |||
| Protects against oxidative and endoplasmic reticulum stress | ||||
| miR-204 | ↓ | SHP2 (Stat3) | Kidney protection against hypertension and diabetes | 84 |
| Prevents renal fibrosis | ||||
| miR-21-5p | ↑ | ERK/MAPK | Inflammation | 85–87 |
| PTEN | Fibrosis | |||
| PPAR-α | Increased oxidative stress | |||
| Smad7 | ||||
| RECK | ||||
| DDAH1 | ||||
| Mpv17 |
ARNT: aryl hydrocarbon receptor nuclear transporter; ATE: atherosclerosis; BCL2: B2 cell lymphoma; VSMC: vascular smooth muscle cells; eNOS: endothelial nitric oxide synthase; CKD: chronic kidney disease; HIF-1α: hypoxia-inducible factor 1α; KLF4/5: Kruppel-like transcription factor 4/5; Mϕ: macrophages; PDCD4: programmed cell death protein 4; PTEN: tensin phosphatase homologue; TNF-α: tumor necrosis factor α; VEGF-A: vascular endothelial growth factor-A.
The role of miR-21 in CKD is significant. It has been observed that miR-21-5p is overexpressed in patients with CKD, and that its levels correlate negatively with glomerular filtration rate.86 Furthermore, miR-21-5p inhibits Smad7, inhibitor of the TFG-β1/Smad3 pathway, so this pathway is enhanced, which generates inflammation and renal fibrosis in these patients. Animal experiments also suggest that miR-21 is also an inhibitor of the Mpv17 protein, responsible for reducing the production of ROS in the mitochondria, so that the increase of this miRNA in the kidney generates an increase in oxidative stress.87
Another important miRNA is miR-155. The absence or downregulation of miR-155 in monocyte-derived EVs in a uremia model generates immune alterations. This is because miR-155 is mainly involved in the maturation of B lymphocytes, the regulation of the adaptive immune response and the control of inflammation, since it is expressed in activated B and T lymphocytes, monocytes, macrophages and dendritic cells.78 These immune alterations could be related to the inflammation and vascular damage observed in patients with CKD since its expression is very important in shaping the transcriptome of active myeloid and lymphoid cells. The reduction of miR-155 has also been associated with osteoclastogenesis and the development of vascular calcification.79
Aging and cellular senescence: mechanism involved in endothelial and vascular damage in CKDAging is defined as a functional decline of the organism that usually involves morphological and physiological changes that entail a loss of homeostatic reserves.88 There are two types of aging: physiological aging and premature or accelerated aging associated with pathologies. Physiological aging is the consequence of time passing by and is therefore closely related to age. When aging appears early and together with the development of diseases associated with aging, it can be identified as premature aging associated with diseases. Thus, renal patients show accelerated aging, since many of them suffer ahead of time diseases associated with aging, such as CVD.4,18,89 Premature aging in these patients can be associated with the accumulation of senescent cells, those that have lost their ability to proliferate. Cellular senescence is defined as a process that takes place as a consequence of cell damage caused by multiple reasons (oxidative stress, proinflammatory cytokines and/or EV), the end result of which is that cells lose their ability to divide, do not die and contribute to the development of age-associated diseases. Among other changes, they are cells that present increased ROS, accumulation of dysfunctional mitochondria, decreased condensation of constitutive heterochromatin with loss of lamin β and increased β-galactosidase lysosomal enzyme activity.88 In addition, senescent cells show a deficit in protein synthesis, as well as an increase in their degradation. Obviously, an increase in senescent cells in a tissue triggers tissue dysfunction.
In patients with CKD, cellular senescence occurs in response to the damage generated by the action of the uremic toxins accumulated during the progression of the disease.90,91 The accelerated cellular senescence attributed to uremia generates an increase in oxidative stress that in turn causes deterioration of cellular structures. As a result, intracellular signals are produced that cause the cell to acquire a senescent phenotype.48 At the systemic level, senescent cells release pro-inflammatory factors that, if persisted over time, generate a generalized deterioration of the organism. This deterioration favors the appearance of tumoral processes and degenerative and chronic illnesses.92
The renal cells are responsible for producing the Klotho protein, an enzyme capable of hydrolyzing the beta-glucurinide steroid. It is a protein with an anti-aging effect, capable of modulating stress-induced senescence and functional response. The decrease in intracellular levels of Klotho has been associated with endothelial senescence, while exogenous Klotho prevents cellular senescence by inhibiting the increase in oxidative stress induced by uremia, as well as inactivating NF-κB and inhibiting its ability to bind to DNA. In this way, the NFκB/IκB complex is stabilized and the release of cytokines that participate in inflammatory processes is reduced. When the levels of this protein are reduced due to uremia, there is an increase in oxidative stress and endothelial senescence is greater.93,94 It has been observed that the α-Klotho protein plays a fundamental role in the maintenance of the integrity and homeostasis of the endothelial cells. Specifically, the functions that have been attributed to α-Klotho are the suppression of the expression of adhesion molecules such as ICAM and VCAM, the attenuation of the NF-κB signaling pathway, and the prevention of hyper-permeabilization95 and apoptosis of endothelial cells by mediating the internalization of the complex formed by the canonical transient potential receptor 1 (TRPC1) and the vascular endothelial growth factor receptor 2 (VEGFR2).96 Patients with CKD present a deficiency of this protein which is due to multiple factors.19 This deficit could explain, at least in part, the greater risk of these patients of suffering CVD. In addition, it has been demonstrated that Klotho, together with the fibroblast growth factor 23 (FGF23) is essential in the axis bone-kidney, since Klotho maintains the homeostasis of the phosphate, collaborating in some way with FGF23 by optimizing the binding of FGF23 to its receptor (possibly it should be under one same route of signaling inhibiting the reabsorption of phosphate), since the degradation of FGF23 leads to a deterioration of the renal function with hyperphosphatemia.94
Finally, it should be emphasized again that endothelial damage is a previous step in the development of several CVDs. The CVD associated with the CKD is partly determined by a rigidity of the blood vessels caused by a state of inflammation and a loss of balance between the pro-aging and anti-aging systems, which finally triggers a process of vascular calcification. At the vascular level, it is observed that patients with CKD present an aged vascular tissue, similar to that found in healthy individuals of advanced age.64
SASP and SIPS in cellular senescence in patients with CKDWhen the cells reach a state of senescence they undergo changes in their phenotype. The secretory phenotype that a cell acquires when it enters senescence is called senescence-associated secretory phenotype (SASP). The secreted material includes mainly chemokines, proinflammatory cytokines (IL-1, IL-6, TNF-α, transforming growth factor β (TFG-β)) and proteases, in such a way that when senescent cells accumulate there is one great release of these compounds that generate a persistent low-level inflammation. In an attempt to neutralize this situation, the cytokines and chemokines will attract the cells of the immune system, mainly monocytes and macrophages, which will eliminate these senescent cells.91 In aging individuals, senescent cells accumulate because there is a greater number of cells undergoing senescence and because their elimination will be slower, since the immune system of these individuals is also senescent. As senescent cells release pro-inflammatory factors, the aging individual has a low-level of chronic inflammation.97 The NF-kB transcription factor, p38 mitogen-activated protein kinase (p38 MAPK) and inflammation are important molecular effectors in the development of this phenotype. Furthermore, IL-1 and TGF-β generate an autocrine positive feedback loop that promotes this proinflammatory situation.90,91 In addition, according to the results obtained in a murine model, senescent T cells release an outburst of cytokines that can cause accelerated senescence of other cells and tissues.98
TGF-β also plays a prothrombotic effect that correlates with aging.99 The stimulation of the renal tubule epithelial cells with TGF-β generates an increase in the production of ROS and nicotinamide adenosine dinucleotide phosphate oxidase (NADP oxidase), which causes vascular aging and the reduction of telomeric length, as well as activation of route p53/21.89,100 Under normal conditions, VEGF and TGF-β together with other factors such as EGF (epidermal growth factor), participate in the healing of wounds by inducing the differentiation of myofibroblasts and increasing collagen deposits, but in advanced stages of this process,101,102 senescent fibroblasts together with TGF-β will generate an excessive fibrosis and an abnormal epithelial-mesenchymal differentiation that correlates with nephropathy. In addition, the TGF-β produced by the podocytes combined with the ROS of the glomerular endothelial cells causes a segmentation of the glomerular capillaries, which generates a renal failure and massive proteinuria in the patient.103
In renal patients there is a so-called SIPS phenotype (stress-induced premature senescence), which differs from the SASP phenotype because SASP is produced by natural aging, whereas SIPS is a consequence of damage or stress, in this case mainly because of the effect of uremic toxins.99,104
Extracellular vesicles in cellular senescenceCellular senescence –triggered either by replication or by other stimuli, such as uremic toxins for example–, considerably increases the secretion of exosomes and MV. As a result, the regenerative capacity of the vasculature is altered, especially that of the endothelial cells, which reduces their capacity for cell migration and their potential to form vascular structures.104 The amount of EV appears modified in multiple pathologies, including the CKD, and therefore its possible role as a biomarker predictor of diseases progression is being evaluated. As said, in CKD patients has been described an increase EV, which is associated with endothelial dysfunction and persistent inflammation.105
Not only is the number of EV altered in renal patients, but also their content, which generates processes of endothelial dysfunction, fibrosis and vascular calcification.106,107 It has been described that one of the triggers of vascular calcification is the senescence of the cells of the vasculature, and is associated with changes in the expression of certain miRNA, as is the case of miRNA-146b-5p and miRNA-223-3p, and these miRNA can be transported in EV.40
Vascular calcification in chronic kidney diseaseThe most prevalent vascular alteration in patients with advanced CKD is vascular calcification, and in many cases it is not detected until the damage is irreversible. Vascular calcification is due to the increase in bone morphogenic proteins and the presence of a high calcium content. The increase in Ca and P in plasma augment the expression of proteins involved in bone formation such as osteopontin, osteocalcin, certain proteoglycans and BMP-2, as well as other factors involved in vascular calcification.12 There are two types of calcifications: 1) calcification of the tunica media, also known as Mönckeberg sclerosis, which affects the VSMC and elastic fibers, which harden and lose their capacity of distension,108 and 2) atherosclerotic calcification of the tunica intima, which is associated with deposition of lipid and lipoprotein followed by calcium accumulation under the tunica intima.109 This deposition can stimulate the development of both innate and adaptive immune responses, inducing the expression of pro-inflammatory molecules in both endothelial cells and VSMC, which stimulates the infiltration of monocytes/macrophages into the tissues. Both types of vascular calcifications are very common in ACKD and may coexist in the same vessel.
The hyperphosphatemia and hypercalcemia are two of the main promoters associated with the development of vascular calcification in the CKD.14,108 The development of calcifications is promoted in patients receiving treatment with vitamin D together with oral calcium salts and who have a positive calcium balance during the dialysis session.110 In vitro models have shown that the increase in phosphorus favors the transformation of VSMC into osteogenic cells, with production of extracellular matrix and subsequent mineralization. Therefore, hyperphosphatemia and hypercalcemia not only passively generate deposits, but also intervene in a highly regulated process by which VSMC differentiate into osteoblasts.111 Eventually, the decrease in the renal function causes blood accumulation of phosphate which, together with the deficiency of circulating inhibitors of calcification and the poor local production of these inhibitors, favors calcification through the activation of Toll-like receptor 4 (TLR-4), which in turn stimulates the expression of NF-κB in VSMCs and leads to an increase in the release of proinflammatory cytokines.14,112
In vitro studies have shown that uremic toxins derived from guanidine (asymmetric dimethylarginine (SDMA), guanidinobutyric acid (GAB), guanidine (G) and guanidino acetic acid (GAA)) are capable of stimulating osteoclastogenesis.113 Uremia also favors the secretion of the osteoblast differentiation factor, Cbfa1, together with an increase in the expression of osteogenic proteins.107,114 In VSMCs the IS stimulates proliferation and favors the production of TGF-β which participates in fibrosis processes.115 Also, the increase in oxidative stress –which is observed in patients with CKD– is closely associated with the development of vascular calcification mediated by the expression of Runx2 (runt-related transcription factor 2) in VSMCs.108 Therefore, various stimuli, such as increased ROS, proinflammatory chemokines and cytokines (CCL5, CCL2, TNF-α, IL-6), and high phosphorus and calcium levels, will facilitate the entry of NF-kB into the core of VSMCs, which stimulates the production of factors involved in calcification (Runx2, OPN, Msh homrbox protein 2 (MSX2), alkaline phosphatase, BMP2) (Fig. 4). In addition, calcium helps to release of EVs, which reinforces the calcification process.107,115–117 VSMCs themselves release a greater amount of EV in response to stress induced by ambient calcium, which also favors vascular calcification.118
In addition, senescence also plays a key role in the development of vascular calcification. It has been observed that EVs released by macrophages of aged individuals participate in the calcification of the vascular matrix.104 Endothelial MVs found in plasma of elderly subjects and released by senescent endothelial cells also help in the calcification of VSMCs, since they contain high levels of calcium, phosphates molecules and proteins that participate in the initiation of calcification.12
Thus, abnormalities in minerals, uremia and the increase in oxidative stress, inflammation and EV release in patients with CKD favor the development of vascular calcification, thereby increasing the risk of suffering cardiovascular damage
Extracellular vesicles in the progresión of cardiovascular illness in patients with chronic kidney diseaseDue to the increase in the prevalence of CKD, it is necessary to identify new therapeutic targets that can slow or reverse the progression of the disease.119 The EV have been proposed as biomarkers for prediction and early diagnosis of CKD. EVs promote alterations in intercellular signaling and vascular damage with the subsequent development of CVD.11,40 Thus, in the presence of abnormal intercellular signaling, there are targets that could be identified that may help to develop methods of early diagnosis and monitoring for patients with CVD associated with CKD.40
EVs, as intercellular messengers, can interact with different cell types through membrane receptors or by being internalized by the target cell through different mechanisms (membrane fusion, endocytosis, phagocytosis, micropinocytosis). In the end, the result of this interaction between EVs and cells is the activation or regulation of different signaling pathways in the target cell.120
Endothelial microvesicles in the development of cardiovascular diseases: endothelial dysfunctionUremic toxins, among which IS and p-cresol stand out, cause damage of endothelial cells, generating a proinflammatory state. This continuous damage of endothelial cells stimulate the release of a greater amount of endothelial MV, which in turn modify vascular tone and further increase vascular damage, thus generating a vicious cycle.11,59,121–123 These endothelial MV are involved in the pathogenesis of several CVD, including atherosclerosis.11,124 There is a general relationship between renal function and cardiovascular risk, which is reflected in excessive production of endothelial vesicles that could act as a marker of endothelial dysfunction.
It has been described that endothelial MV promote vascular calcification, since treatment of VSMC with endothelial VM from endothelial cells previously treated with IS induced calcification in VSMC, whereas endothelial MV from endothelial cells not treated with uremic toxins did not produce calcification.107
Platelet microvesicles in the development of cardiovascular disease: coagulationPlatelet MV are another type of MV implicated in the development of CVD. In this pathological process there is excessive platelet activation which is followed by platelet aggregation, secretion of their internal granules and release of a greater number of MV, all of which have procoagulant activity.120 Moreover, in a situation of endothelial dysfunction, platelets tend to adhere to the damaged surface,125 with the consequent release of potent mitogenic factors leading to the proliferation of VSMs and the progression of vascular damage.
The dialysis technique itself may be a risk factor for renal patients. There are several studies that show hypercoagulation in these patients, probably due to increased expression of tissue factor (TF).126 TF, also known as thromboplastin, is expressed in the cell membrane and is synthesized by different cell types; and, it can be found on the surface of monocytes and endothelial cells in inflammatory states. This factor is involved in local thrombi formation. TF is expressed on the surface of EV, so it can bind to the platelet surface of an evolving thrombus, which occurs frequently in uremic patients.127 Likewise, it has been shown that elevated levels of TF, important in thrombogenic activity, can be crucial in patients undergoing some types of RRT.11 This suggests that EVs expressing TF may be biomarkers of thrombotic events in uremic patients.127
Extracellular vesicles as a diagnostic biomarkerIn recent years, the analysis of EV in body fluids has been used as a diagnostic tool (biomarker) in various pathologies, this is the case of patients with CVD associated to CKD.40,128 Biomarkers are an analytical tool that provides objective, measurable and evaluable information on biological function, some pathological processes or the response to treatment. Biomarkers are essential in clinical practice because they allow early identification of a given pathology, and thus allow early therapeutic intervention that prevent the development of such pathology and the possible negative consequences for the individual.11 In general, high levels of EV have been detected in numerous pathologies, particularly those of a chronic and inflammatory nature, such as diabetes, hypertension, cancer and CKD.129
In the context of CKD, a relationship has been observed between renal function and cardiovascular risk, all of which is reflected in an elevated production of endothelial vesicle that could act as a marker of endothelial dysfunction.105 Specifically, Mezentsev et al. described that the physiological levels of endothelial MV in healthy individuals is between 103–104MV/mL, while in individuals with CVD it is 105MV/mL.130 Different studies point to the existence of a higher concentration of circulating EV in patients with CVD such as congestive heart failure, coronary artery disease, peripheral vascular disease and cerebral ischemia.60
In uremic patients there are also high levels of platelet MV with a procoagulant effect, which can be associated with coagulation disorders, cerebrovascular accidents, unstable angina and acute myocardial infarction. This explains its possible contribution to the development of CVD (especially associated with thrombosis and platelet instability) and its use as a possible diagnostic biomarker.131,132 The levels of platelet MV in patients on HD and PD were higher than in healthy people; and even more increased than in those patients who suffered a thrombotic event.131 It has also been observed that patients with CKD who have suffered an acute myocardial infarction – and therefore with a high risk of suffering a new thrombotic event – have a high levels of platelet MV and that the levels of platelet MV are higher in those patients with a more advanced CKD.133 These platelet MVs express P-selectin, which indicates platelet activation. In addition, as mentioned above, the increase in VE with TF can be a clinical marker of risk of thrombotic event in patients with CKD.127
Extracellular vesicle-based therapiesIt has been proposed that EVs and their content may act as therapeutic targets. Firstly, since CVD has been related to the increased release of EVs, pharmacological strategies can be designed to inhibit the release EVs and even their interaction with the target cells. For example, ceramide which is an important component in exosome biogenesis can be inhibited.63 Even so, treatments focused on the inhibition of EV release are scarce, due to the lack of specific knowledge of the mechanisms involved in the biogenesis and release of EVs.11 Treatments focused on reducing EV release could reduce the morbidity and mortality of patients with CKD.63
It has been observed that some drugs that have been used traditionally in the treatment of CVD and CKD modulate the release of EV (Table 3).
Treatments that reduce the release of extracellular vesicles into the blood.
| Compound | Type of EV reduced | Pathology | Reference |
|---|---|---|---|
| Statins | Endothelial | Hypertension | 134–136 |
| Platelet | Diabetes mellitus type 1 | ||
| Leukocyte | |||
| Simvastatins+losartan | Endothelial | Hypertension | 135 |
| Platelet | Diabetes mellitus type 2 | ||
| Monocyte | |||
| Aspirin | Endothelial | Coronary heart disease | 11,129 |
| Platelet | |||
| Antihypertensives | Endothelial | Hypertension (animal model) | 137 |
| Antioxidants | Endothelial | Diabetes mellitus type 1 | 11,129 |
| Platelet | Dyslipemia |
Studies have been performed to test whether endothelial MV levels could be modulated by drugs. For example, the use of statins, specifically atorvastatin, produced a decrease MV and VEGF levels.134 This is possible because statins inhibit the Rho-kinase pathway which is involved in skeletal reorganization and MV release, thus the release of MV is reduced (not only endothelial MV but also those derived from platelets and leukocytes).135,136 Furthermore, in patients with hypertension and type 1 DM, treatment with simvastatin together with losartan has been observed to decrease plasma concentrations of endothelial, platelet and monocyte-derived MV.135 Aspirin could also have a reducing effect on EV release, specifically on endothelial and platelet, but the results are somewhat heterogeneous.11,129 A reduction in peripheral blood endothelial MV was also observed in an animal model of hypertension in which antihypertensive drugs (aliskiren, nerbivolol and olmesartan) were used; this reduction also mediates the antiangiogenic effects of these drugs.137
Antioxidants such as vitamin C could also reduce the release of VE; specifically, in patients with type 1 DM and dislipemia after suffering a myocardial infarction.11
In addition to using EVs as a target for treatments, it has recently been investigated the use of EVs as a therapeutic tool, as a way to deliver a treatment in a more effective and “physiological” manner. This idea arises from the fact that EVs under physiological conditions may have beneficial effects on target cells. For example, it has been observed that EVs may be involved in tissue repair and immune modulation processes, therefore they could be used directly as therapeutic agents in regenerative medicine and the treatment of autoimmune diseases.63
Under physiological conditions, platelet-derived MVs are capable – through VEGF – of stimulating proliferation, survival, cell migration and the formation of new capillaries. Additionally, the endothelial MV are related with to regenerative processes and the maintenance of vascular homeostasis; therefore, an increase in the levels of this type of vesicle is key in processes where vascular regeneration is taking place.11,120
Regarding the kidney, models of diabetic nephropathy, CKD, fibrosis and acute kidney injury, have shown that EV derived from mesenchymal stem cells have a renoprotective function. In general, EVs derived from stem cells are the ones that present the greatest beneficial effects in kidney diseases. This could be due to its content rich in miRNAs and proteins (mainly related to cell proliferation, migration and adhesion).63
Finally, the use of EVs as a vehicle for treatments (which could be drugs, miRNAs or proteins) has also been explored. This type of delivery has the advantage that they are less immunogenic and cytotoxic, and up to now the evidence is that they seem to have less rejection than nanoparticles.63
ConclusionsPatients with CKD, due to uremia, present an increase in inflammation and oxidative stress and, consequently, premature aging, with accumulation of senescent cells, which explains the early onset of diseases associated with advanced age, such as CVD. The antecedent of CVD, so prevalent in renal patients, is the endothelial damage caused by oxidative stress, chronic inflammation and EVs released as a result of cell damage. These EVs contain miRNAs, such as miR-21 and miR-155, which alter the expression of different proteins and may be responsible for the increase in vascular permeability and the maintenance of inflammation.
Patients with CKD present a greater number of EVs in plasma; in particular, endothelial MVs stand out, which are increased due to endothelial damage in these patients and represent a first step towards more severe vascular complications such as vascular calcification. The endothelial MV of patients with CKD have a high content of calcium and calcium-binding proteins that facilitate or promote vascular calcification and, consequently, can trigger CVD. In these patients there may also be alterations in coagulation, a consequence of endothelial dysfunction and platelet MV that appear in greater quantities in the plasma of uremic patients. Platelet MV could also participate in the formation of atheroma plaques and vascular calcification.
With this background, the measurement of MV can be a good predictor of cardiovascular complications in patients with CKD, enabling early detection and facilitating an early therapeutic approach. Even so, thresholds have yet to be established above which it would be possible to predict whether there is an increased risk of CVD and transfer these patients to the clinical setting. EVs could also be a good therapeutic target to slow down the development of CVD in the early phases of the disease, but this requires further research into drugs that modulate the release of EVs. Nevertheless, there are already known drugs commonly developed to treat CVD or its risk factors that modulate EV release; and thus have a beneficial effect on these treated patients. It has also been shown that EVs released mainly by stem cells have a content that can be beneficial for the individual, and could therefore be of interest for treating patients with CKD who are highly predisposed to suffer CVD. It has been proposed the use of EVs as a vehicle to target drugs of a given treatment in a more specific way.
In summary, the use of EVs as markers for the prediction, diagnosis, prognosis and monitoring of therapies for complex diseases is becoming increasingly attractive, as is their potential for identifying new therapeutic targets. In this regard, clinical and experimental evidence suggests that EVs may be useful in monitoring the efficacy of replacement therapies; indeed, the results obtained to date in the various studies are very promising.
FundingThis work has been funded by the Instituto de Salud Carlos III through project PI17/01029, PI19/00240, and PI20/01321 (co-funded by the European Union (ERDF/ESF) “Una manera de hacer Europa”/“El FSE invierte en tu future”), and the Spanish Society of Nephrology (Senefro Foundation Project). AF is a predoctoral fellow of the program: “Contratos Predoctorales de Investigación en Salud, Instituto de Salud Carlos III (FI20/00018)”. GV received a grant from the Community of Madrid and the European Social Fund (PEJ-2020-AI/BMD-18141). NS has a grant from the program Grants Associated with Research Projects, Instituto de Investigación Sanitaria Hospital 12 de Octubre (i+12), Madrid, Spain.
Conflicts of interestThe authors do not declare a conflict of interest.



