




Although phosphorus is an essential element for life, it is not found in nature in its native state but rather combined in the form of inorganic phosphates (PO43−), with tightly regulated plasma levels that are associated with deleterious effects and mortality when these are out of bounds. The growing interest in the accumulation of PO43− in human pathophysiology originated in its attributed role in the pathogenesis of secondary hyperparathyroidism (SHPT) in chronic kidney disease. In this article, we review the mechanisms by which this effect was justified and we commemorate the important contribution of a Spanish group led by Dr. M. Rodríguez, just 25 years ago, when they first demonstrated the direct effect of PO43− on the regulation of the synthesis and secretion of parathyroid hormone by maintaining the structural integrity of the parathyroid glands in their original experimental model. In addition to demonstrating the importance of arachidonic acid (AA) and the phospholipase A2-AA pathway as a mediator of parathyroid gland response, these findings were predecessors of the recent description of the important role of PO43- on the activity of the calcium sensor-receptor, and also fueled various lines of research on the importance of PO43− overload not only for the pathophysiology of SHPT but also in its systemic pathogenic role.
Aunque el fósforo es un elemento indispensable para la vida, en la naturaleza no se encuentra en estado nativo sino combinado en forma de fosfatos inorgánicos (PO43−), con niveles plasmáticos estrechamente regulados que se asocian a efectos deletéreos y mortalidad cuando estos se encuentran fuera de la normalidad. El interés creciente sobre el acúmulo de PO43- en la fisiopatología humana se originó en el papel que se le atribuyó en la patogenia del hiperparatiroidismo secundario a la enfermedad renal crónica. En este artículo revisamos los mecanismos por los cuales se justificaba dicho efecto y conmemoramos la importante contribución de un grupo español liderado por el Dr. M. Rodríguez, ahora hace justo 25 años, cuando demostraron por primera vez el efecto directo del PO43− sobre la regulación de la síntesis y secreción de hormona paratiroidea, manteniendo la integridad estructural de las glándulas paratiroides en su nuevo modelo experimental. Además de demostrar la importancia del ácido araquidónico (AA) y la vía de fosfolipasa A2-AA como mediadora de respuestas en la glándula paratiroidea, estos hallazgos fueron predecesores de la reciente descripción del importante papel del PO43- sobre la actividad del receptor-sensor de calcio y alimentaron asimismo diversas líneas de investigación sobre la importancia de la sobrecarga de PO43− no sólo en la fisiopatología del HPTS sino también en su papel patogénico sistémico.
Phosphorus (P), (in Greek phos phorus or carrier of light), is an essential element (atomic number 15, molecular weight 30.9 u) found in all living organisms. It has a dual function: (a) structural; in nucleic acids (DNA and RNA), cell membranes (phospholipids) and in the mineral phase of bone (together with calcium it forms hydroxyapatite crystals); (b) regulatory; constituting the main energy intermediate in cellular processes such as metabolism and activation of proteins (phosphorylation), involved in the oxygen dissociation curve of hemoglobin (2,3-diphosphoglycerate) and in cell signaling processes (second messengers such as cyclic AMP and GMP). In nature, including living organisms, phosphorus is not found in its native state, but mainly combined in the form of various non-flammable inorganic phosphates (PO43–) (Fig. 1), this being the form in which phosphorus levels in the body are measured. Its biological importance determines that PO43– levels must be strictly controlled within an optimal physiological range and that outside of these levels it is associated with deleterious effects. Thus, multiple epidemiological studies show not only that iPO43– deficiency is a cause of pathology, but also elevated PO43– levels (even in the normal ranges) have been independently associated with cardiovascular disease and mortality in the general population and, especially in patients with chronic kidney disease (CKD), whether on dialysis or not.1–7
Considering that kidneys play a fundamental role in the regulation of PO43–, alterations in its homeostasis in CKD are to be expected because the balance depends on its intake and excretion. The intra- and extracellular accumulation of PO43– plays an essential role not only in the pathogenesis of secondary hyperparathyroidism (SHPT) and the chronic kidney disease-mineral and bone disorder (CKD-MBD) complex, but also, directly or indirectly, it can be considered as a (non-classical) risk factor for cardiovascular and even infectious morbidity and mortality, through various mechanisms including inflammation, oxidative stress, vascular and valvular calcifications or myocardial fibrosis. It also seems to contribute to cellular dysfunction of the immune system, arteriosclerosis-atheromatosis, and even accelerated aging in CKD patients.1,8–10 Recently, the role of PO43– and calcium phosphate microcrystals in the renal tubular lumen have also become relevant, not only as a secondary effect but also as a causal factor in the progression of CKD itself.11,12
However, the growing interest in PO43– accumulation in human pathophysiology was originated by the role attributed to PO43– in the aforementioned pathogenesis of SHPT. Therefore, the objective of this article is to review the mechanisms by which this effect was justified, and commemorate the important contribution of a Spanish group to the first demonstration in experimental studies of the direct effect of PO43– on the regulation of synthesis and secretion of parathyroid hormone (PTH) in the parathyroid glands, just 25 years ago now.13
Phosphorus in the pathogenesis of secondary hyperparathyroidism: indirect mechanismsFor many years there has been a profound, almost bipolar, debate about whether the retention of PO43– or the decrease in the synthesis of calcitriol (1,25-[OH]2-vitamin D), both present in patients with CKD, were the initiating or most important factors in the pathogenesis of SHPT.14–16 Exactly five decades ago now (golden anniversary), Slatopolsky et al.14 observed a significant increase in PTH in an experimental model in dogs with CKD fed a high PO43– diet. The interpretation of these findings was that a transient increase in postprandial PO43– would decrease ionized calcium in the blood, and this decrease in calcium would be what would stimulate PTH secretion. The increase in PTH would not only normalize calcium levels, but would also decrease tubular phosphorus reabsorption, with the consequent increase in phosphaturia, thus normalizing the serum levels of both ions at the expense of elevated serum PTH. Subsequent decreases in glomerular filtration (GFR) would lead to a progressive increase in the PTH levels necessary to normalize calcium and PO43–. These observations constituted the “trade-off hypothesis” (or PTH elevation –and its consequent deleterious effects– at the expense of trying to maintaining calcium and PO43– homeostasis)17,18 (Fig. 2). These same authors demonstrated in another experimental study that the proportional reduction in PO43–intake adjusted to the gradual decrease in GFR was able to prevent SHPT, as evidenced by the presence of normal plasmatic levels of calcium, PO43–, PTH and tubular phosphorus reabsorption as compared to controls.19 In this experimental model, the explanation for the observations made was based on the fact that the adaptation of the remaining nephrons to the CKD stage was necessary, in which these nephrons must respond to a greater fraction of renal excretion of PO43– with a decrease of its tubular reabsorption (as usually happens in the unadjusted intake of PO43– or other elements).20 This hypothesis was postulated by Bricker under the so-called «intact nephron hypothesis»20 and the findings then suggested that a constant intake of PO43–in the face of a diminished nephron population (CKD) would play an important role in the pathogenesis of SHPT.20,21
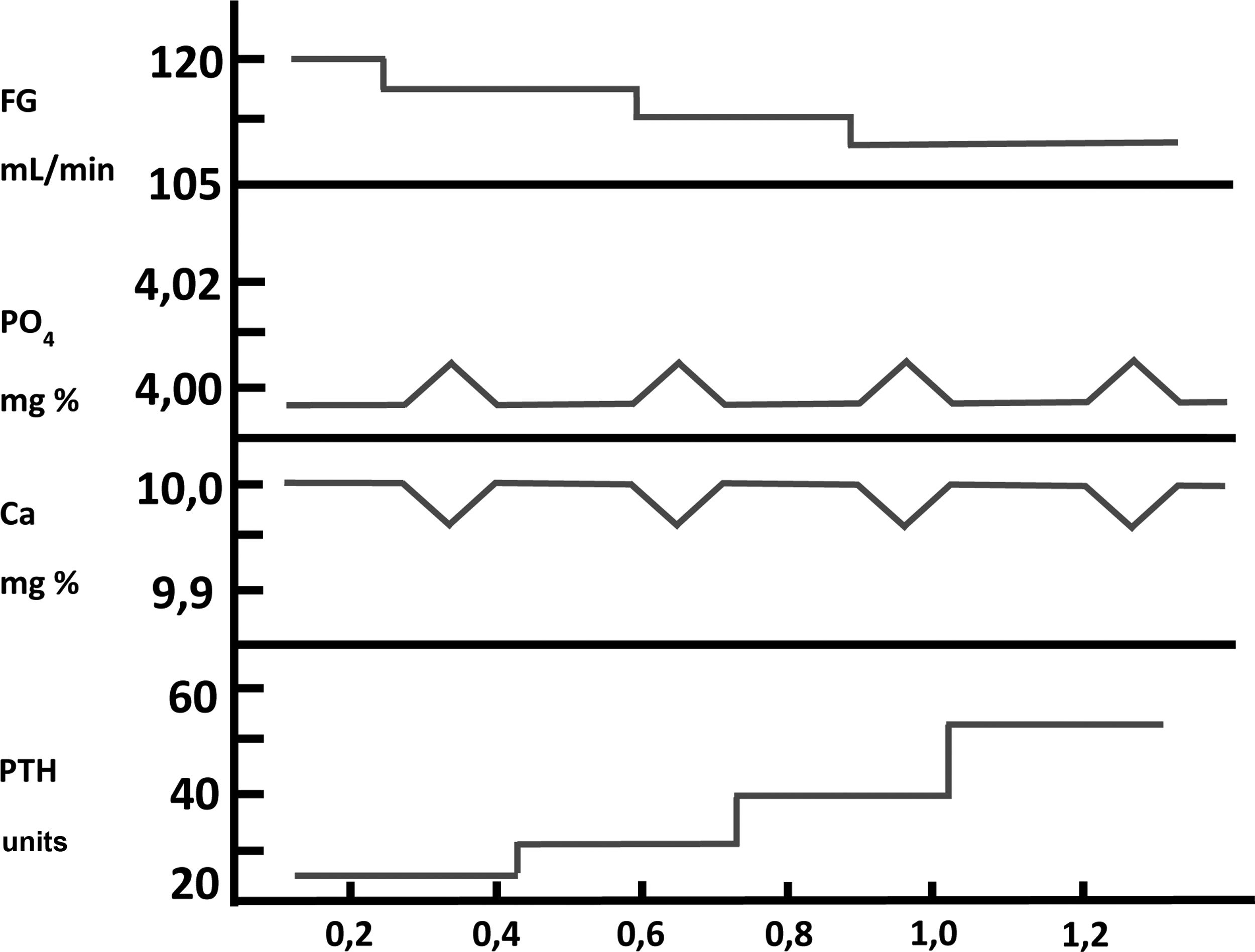
Pathogenesis of secondary hyperparathyroidism in chronic kidney disease. Representation of the « trade-off hypothesis ». (Adapted from Bricker et al.18).
Although in the aforementioned study19 the reduction in PO43– prevented SHPT, the possibility of the contribution of PTH inbibition by an increased calcitriol synthesis induced by the low PO43– diet could not be ruled out. This fact would be particularly relevant, especially in the early stages of kidney damage, as demonstrated by Portale et al.15 years later in children with moderate CKD. PO43– is a known inhibitor of 1 α-hydroxylase (CYP27B1) and in these children it was shown that a diet high in PO43– stimulated PTH and decreased calcitriol levels, whereas a diet low in PO43– stimulated calcitriol with a secondary decrease in PTH. In addition, it is common for a diet low in PO43– to be associated with an increase in serum calcium due to the greater intestinal absorption of calcium secondary to the increase in calcitriol, so that this increase in serum calcium could explain, at least in part, the decrese in PTH. Thus, Llach and Massry16 demonstrated in four patients with moderate CKD that PO43– restriction favored the action and production of calcitriol, modifying not only intestinal calcium absorption but also the calcemic response to PTH, thus suggesting an important role of altered vitamin D metabolism in the pathogenesis of SHPT in CKD.
In those days, the “so called “decreased calcemic response or skeletal resistance to the action of PTH (endogenous and exogenous) was another indirect mechanism described by which PO43– retention and/or hyperphosphatemia could contribute to the development and progression of SHPT in CKD,22,23 and this effct has been widely studied by Rodriguez et al.24–27 Currently called « hyporesponsiveness» to PTH,28 various studies have shown the relative importance of PO43– retention on the resistance to the action of PTH, among other factors.24–30 As reviewed in a recent article in this journal,30 the existence of a hyporesponse to the action of PTH, through various mechanisms, among which it stands out the retention of PO43–, demands a greater synthesis and secretion of PTH to maintain mineral homeostatic balance.
Phosphorus in the pathogenesis of secondary hyperparathyroidism: direct mechanismUntil now, the mechanisms described in this review on the role of PO43– in the pathogenesis of SHPT have been indirect (through a decrease in ionic calcium, a decrease in calcitriol synthesis or multifactorial hyporesponsiveness to the actions of PTH in CKD). It is well known that both the decrease in extracellular calcium concentration and the decrease in calcitriol levels have a direct stimulatory effect on PTH synthesis and parathyroid cell proliferation, mediated by their respective receptors (the calcium sensing receptor [CaSR] and vitamin D receptor [VDR]).31–34 In fact, Sherwood et al.35 failed to find evidence of a direct effect of PO43– on the regulation of PTH in vivo.
However, López-Hilker et al.,36 in another experimental study in dogs with CKD on PO43– and also calcium restriction in order to prevent hypercalcemia hypercalcemia, observed that the serum PTH level decreased even without changes in calcium and calcitriol. These findings suggested that the control of SHPT was independent of plasma calcium and calcitriol levels. Years later, in an experimental model in rats, Yi et al.37 also found evidence of the existence of calcium and calcitriol-independent mechanisms to control PTH and, therefore, a possible direct effect of PO43– on parathyroid function.
On the other hand, since it is difficult to ignore the direct effect of calcitriol on the decrease in PTH synthesis and secretion when a low PO43– diet is prescribed, Kilav et al.38 also demonstrated that second-generation vitamin D-deficient rats fed a diet low in PO43– and calcium had hypophosphatemia associated with low PTH mRNA levels in the absence of hypercalcemia or increased calcitriol levels, suggesting a non-transcriptional effect of PO43–, in contrast to the direct effects of calcitriol by decreasing the transcription of the pre-pro-PTH gene. It has subsequently been shown that this non-transcriptional effect of PO43– is actually post- transcriptional, involving the binding of transactivating proteins (proteins that act on trans or adenosine-uridine-rich binding factor [AUF1]) to cis domains (cis element) located at the 3' untranslated region of PTH mRNA, orchestrated by Pin1 isomerase, ultimately leading to increased PTH mRNA stability.39–42
Finally, in a preliminary work, Hernández et al.43 had also described in a preliminary work the effects of a diet high in PO43– in an in vivo rat model and found, compared to rats that ingested a standard PO43– diet, a significant increase in the levels of PTH associated with a rapid increase in PTH mRNA expression without affecting calcium or calcitriol levels. As they subsequently demonstrated, this observation was not accompanied by changes in the expression of VDR or RSCa,43–45 suggesting that an oral PO43– load could directly stimulate PTH synthesis and that it was not mediated by decreased expression of the calcium or calcitriol receptors.
Then, using a completely original experimental model, based on the maintenance of the structural integrity of the parathyroid gland, a Spanish group led by Dr. M. Rodríguez described for the first time in vitro and in an unquestionable manner the direct effect of PO43– on PTH secretion (IX Latin American Congress of Nephrology in San Juan, Puerto Rico, October 20–23, [1994] and the XIIIth International Congress of Nephrology in Madrid, July 2–6, [1995]). Thus, Almaden et al.13 published in 1996 their work in which they used whole fresh rat parathyroid glands that were incubated with different concentrations of PO43– (1, 2, 3, and 4 mM) and were subsequently exposed to different calcium concentrations in a range from 0.4 to 1.35 mM. The authors found that at a calcium concentration of 1.25 mM, PTH secretion was similar with PO43– concentrations of 1 and 2 mM; however, PO43– concentrations of 3 and 4 mM produced an increase in PTH secretion three and four times greater, respectively, as compared to PO43– of 1 mM (Fig. 3). Likewise, in concentrations of 1 or 2 mM of PO43– an increase in calcium concentration from 0.6 to 1.35 mM reduced PTH secretion to 37%; whereas in PO43– of 4 mM the same increase in calcium concentration inhibited PTH secretion only to 75% (Fig. 3). This pioneering demonstration of the essential role of the structural integrity of the parathyroid gland and the maintenance of cell-cell contacts was a fundamental starting point to accelerate the progress of knowledge on the impact of PO43– in the pathogenesis of SHPT in different laboratories around the world. In fact, until then, with the same objective of evaluating the possible direct effect of PO43– on parathyroid function, dispersed parathyroid cells were used in in vitro studies showing a lower response to changes in extracellular calcium than in the intact parathyroid gland model used by Almadén et al.13 The fact is that in isolated (dispersed) parathyroid cells the response to changes in extacellular calcium is reduced due to a decreased expression of the RSCa.46
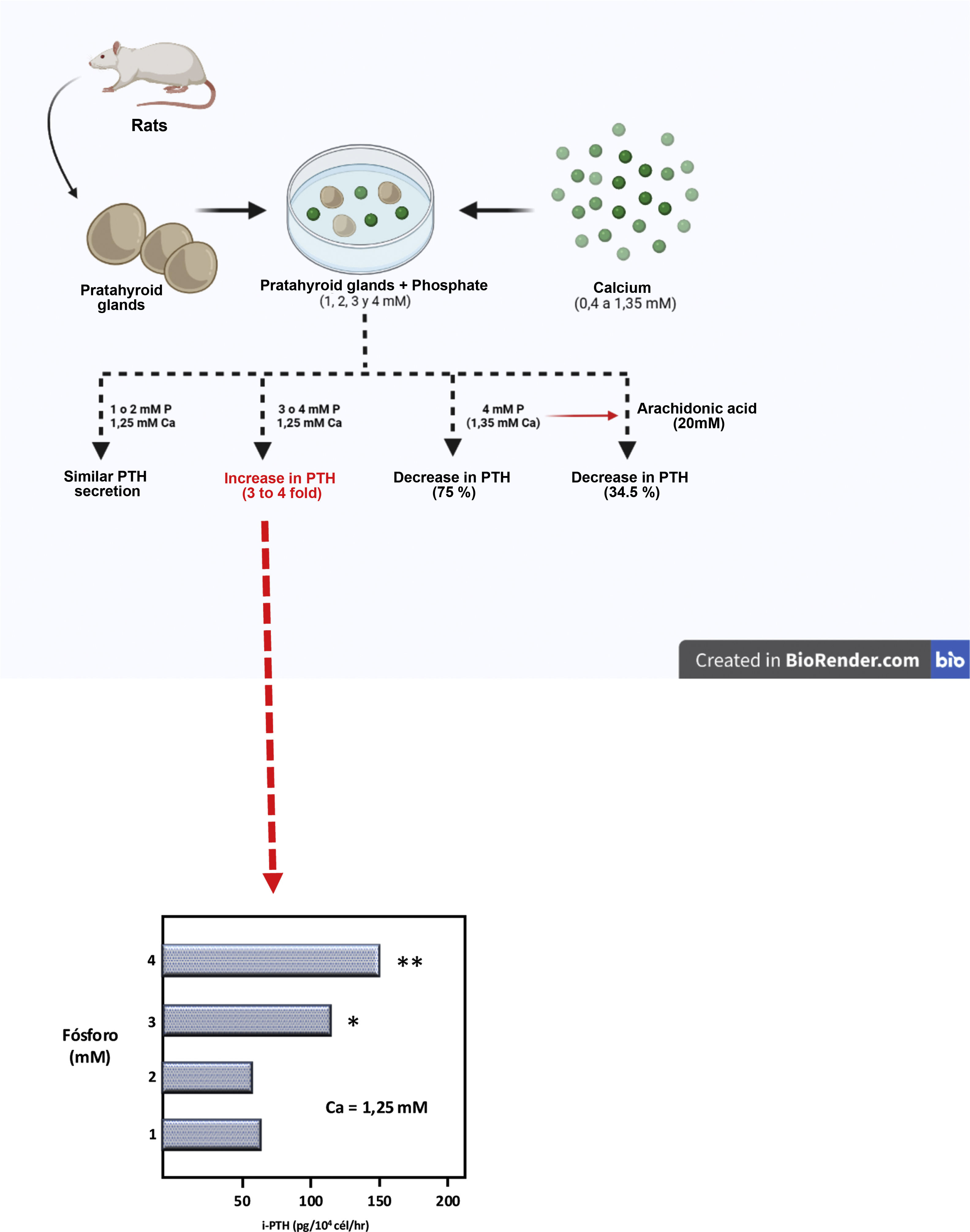
Theoretical representation of the model used by Almadén et al.13 for the demonstration of the direct effect of phosphorus (P), independent of calcium (Ca), on the stimulation of parathyroid hormone (PTH) secretion in the parathyroid gland. The glands were incubated for 1 h in a calcium concentration of 1.25 mM and a variable phosphorus concentration of 1, 2, 3 and 4 mM.
In this seminal study by Almadén et al.,13 the authors also studied the intracellular signaling mechanisms that mediate the direct effect of PO43– on PTH secretion. Thus, the addition of arachidonic acid (AA, a substrate that inhibits the intracellular signaling pathway), to the incubation medium of PO43– 4 mM and calcium 1.35 mM reduced PTH secretion to 34.5%. The conclusion of the study was therefore that in this model using fresh whole rat parathyroid glands, the elevation of PO43–in vitro directly increased PTH secretion by acting through the phospholipase A2 (PLA2)-AA pathway.
Subsequently, they also demonstrated in vitro that, in glands from patients with severe SHPT, the PTH secretion increased in response to PO43– 3 and 4 mM as compared to 2 mM despite the presence of a high concentration of calcium in the medium; this effect was accompanied by an increase in pre-pro-PTH.47 Likewise, this same group demonstrated that an acute elevation of serum PO43–, without changes in the ionic calcium concentration, also stimulated PTH secretion in vivo.48 They also showed the effect of high PO43– concentrations on AA production in parathyroid tissue in vitro,49 and the regulation of AA production by intracellular calcium,50 showing that the stimulation of PTH secretion by high PO43– levels could be prevented by increasing intracellular calcium levels. They also reviewed the importance of AA and the phospholipase A (2)-AA pathway as a mediator of responses in the parathyroid gland.51 In addition, it was shown that maintaining elevated serum PO43– levels during the construction of calcium-PTH curves in hemodialysis (using high or free PO43– dialysis fluid) partially prevented the inhibition of PTH secretion by calcium (normal or elevated).52
On the other hand, from the laboratory of Slatopolsky et al. (J of Investigative Medicine, April [1995]), using the same experimental model of intact parathyroid glands as used by Almadén et al.,13 it was publsihed in the same year, 1996, the effect of dietary PO43– on PTH levels, PTH mRNA, and parathyroid hyperplasia in uremic and normal rats.53 The authors observed that gland weight and serum PTH were similar in both groups exposed to a low PO43– diet (0.2%), but there was a significant increase in serum PTH, gland weight, and DNA in the parathyroid glands from uremic rats fed a high PO43– diet (0.8%) compared with uremic rats fed a low PO43– diet. Additionally, they observed that the stimulatory effect of extracellular PO43– on PTH production did not occur when protein synthesis was inhibited with cycloheximide, suggesting that the action of PO43– on parathyroid cells required protein synthesis. Likewise, the growth rate of parathyroid cells was independent of calcium and calcitriol levels. Previously, Denda et al.54 had already shown that the effect of PO43– on the growth of parathyroid glands was very rapid in uremic rats, two months after the induction of renal failure, and that 90% of the growth occurred during the first three days, potentially attributed to the participation of proto-oncogenes such as c-fos, c-jun and PRAD-1.
Interestingly, in the same year, acknowledging the work of the two groups led by doctors Rodríguez and Slatopolsky, and with the same objective of investigating the direct effect of PO43– on parathyroid cells in vitro, Kjaerulff et al. from Dr Olgaard's Danish group,55 used two types of bovine parathyroid tissue preparations: dispersed parathyroid cells and parathyroid tissue sections, both incubated for four hours in normal (1.0 mM) or high PO43– (3.5 mM) and observed a significant increase in the release of PTH in the parathyroid tissue sections incubated in medium high in PO43– but not in the preparation with dispersed cells, without observing changes in the " set-point " of calcium. The degree of stimulation of PTH release with high PO43– in the medium was significantly higher compared with low calcium medium (0.8 mM), 172% above baseline (1.0 mM PO43–) and 139% higher in a high calcium medium (1.8%). Their results also demonstrated that PO43– directly stimulates PTH release in sections of bovine parathyroid glands and not in dispersed cell preparations, confirming that maintenance of normal parathyroid architecture is essential to reproduce the stimulatory effect of increasing PO43– in PTH secretion.
Effect of phosphorus on the parathyroid glandsUnder normal conditions, parathyroid cells are in a quiescent state and rarely undergo mitosis; however, it is well known that in CKD several factors such as hypocalcemia and calcitriol deficiency induce growth of the parathyroid glands, stimulating parathyroid cell proliferation of parathyroid cells, initially at the expense of cell hyperplasia and, consequently, the synthesis and secretion of PTH.56–59 We have already mentioned that a diet high in PO43–, either by direct or indirect mechanisms, promotes parathyroid growth from an early stage, as demonstrated by Denda et al. in experimental studies in rats.54 Conversely, a low PO43– diet and calcitriol administration were also shown to prevent uremia-induced hyperplasia and PTH secretion. Thus, Dusso et al.60 demonstrated in uremic rats that a diet low in PO43– was able to prevent parathyroid gland hyperplasia by increasing p21 protein and mRNA in parathyroid tissue. The protein encoded by the p21 gene is an inhibitor of cyclin-dependent kinases (Cdk complexes) and, therefore, a regulator of the cell cycle. Likewise, this study also demonstrated that a diet high in PO43– induced the expression of transforming growth factor-α(TGF-α) as an autocrine signal that stimulated parathyroid growth. Such elevations of TGF-α in the parathyroid gland induced by hyperphosphatemic diets and the consequent activation of the epidermal growth factor receptor (EGFR) were identified as the determinants of the decrease in vitamin D receptor (VDR) and the origin of resistance to control of HPTS with calcitriol or its analogues with the progression of CKD.61 It is also important to highlight that, as CKD progresses, not only VDR expression decreases, but also the expression of the CaSR and fibroblast growth factor receptor 23 (FGFR) during the evolution from diffuse hyperplasia to nodular hyperplasia.59 More recently, it was also shown that a high demand for PTH secretion, promoted either by a diet very high in PO43– or low in calcium, induced different patterns of parathyroid hyperplasia in the absence of uremia, a situation that could be important in early stages of CKD.62
It is now interesting to note that the regulation of PTH by PO43– also involves certain micro-RNA (miRNA).63,64 The miRNAs are small non-coding RNAs with vital functions in homeostasis and development of the organism, and we know that the Dicer enzyme is involved in the final stage of miRNA processing. In this regard, for example, we have recently learned that parathyroid cell Dicer -specific knockout mice (PT- Dicer -/-) have normal serum PTH levels, but cannot increase PTH in hypocalcemia or renal failure, unlike controls.64 Also, in addition to modulating PTH secretion, miRNAs are essential for keeping intact parathyroid glands. Dicer knockout mice do not express miRNA in parathyroid cells and lose their parathyroid glands after birth. This indicates that miRNAs are not essential for embryonic development of the parathyroid glands, but rather for their integrity during the postnatal period. In the absence of parathyroid glands, in adult PT - Dicer -/- mice, an additional source of PTH, other than thyroid cells or the thymus, contributes to the maintenance of normal serum PTH concentrations, has been demonstrated.but cannot be stimulated by hypocalcemia or an uremic state.64
Other aspects related to phosphorus overload in chronic kidney diseaseIt is not the purpose of this article to review the entire complex pathophysiology of SHPT but, as mentioned, to commemorate the 25th anniversary of the important discovery of the direct effect of PO43– on the parathyroid gland, in which Spanish researchers played such an important role.13,53 However, it is necessary to mention the importance of the discovery of a phostatonin, fibroblast growth factor 23 (FGF23),65,66 a hormone produced by osteocytes and whose production is mainly stimulated by PO43– overload, among other factors.67,68 Today we know that its elevation occurs from early stages of CKD, even before the elevation of serum PTH, which in turn also stimulates the production of FGF23.69–71 FGF23 not only has a phosphaturic action by inhibiting the expression of Na-Pi 2a and 2c channels at the renal tubular level (decreasing tubular reabsorption of PO43– and thus increasing its urinary excretion) but also inhibits renal 1α-hydroxylase (CYP27B1) (responsible for the synthesis of calcitriol) and stimulates 24-hydroxylase (CYP24A1) (increasing its catabolism).69,70 This new counterregulatory mechanism of PO43– overload allows us to revisit the old, but still current, “trade-off” hypothesis17,21,70,72 and the importance of PO43– in the pathogenesis of SHPT in CKD, by providing a new previously unknown mediator, the FGF23. This increase in FGF23 is explained at least in part by the reduced expression of its co-receptor Klotho, which is necessary to exert its action in target tissues (parathyroid, vascular, brain, renal tubules). This has been demonstrated in several studies and in the case of kidney tissue, it will lead to resistance to the action of FGF23.73 It is known that the action of FGF23 is mediated by a canonical affinity between its receptor FGFR and its Klotho co-receptor,10,74 so that by decreasing Klotho production and/or FGFR expression as a consequence of CKD,73–76 it will cause another hormonal hyporesponse (resistance), in addition to that of PTH or other hormones such as insulin or growth hormone.30 This hyporesponse to FGF23 will lead to an additional increase in the levels of FGF23 required to exert its phosphaturic action in the presence of CKD and/or phosphorus overload, but the excces of FGF23 will produce deleterious secondary effects (such as another "trade-off", in this case at the expense of high FGF23).68,73,77–79 In fact, both the decrease in Klotho and the increase in FGF23 typical of CKD have been clearly associated with accelerated aging and disproportionately high mortality in CKD patients, especially in its advanced stages or on dialysis,2,10,80 turning CKD into an unfortunate human experimental model of senescence.8,9 Therefore, the intra- and extracellular retention of PO43– constitutes one of the main stimuli for the synthesis and secretion of both hormones (PTH and FGF23), either directly or indirectly, with the purpose of increasing phosphaturia, among other effects. In addition, PTH and FGF23 act competitively in the enzymatic regulation of vitamin D production and catabolism. This complex hormonal interrelationship is exacerbated by the progressive loss of renal parenchyma as CKD progresses. Furthermore, the secondary reduction of klotho, the increase in PO43– and the increase in FGF23 act as proinflammatory stimuli, being key elements in the inflammatory state consubstantial to CKD and involved in multiple deleterious effects associated with the loss of renal function. (development of anemia and resistance to erythropoiesis-stimulating agents, protein-energy malnutrition syndrome, endothelial dysfunction), as well as early and accelerated calcifications and atherosclerosis.81–83
CorollaryUnder normal conditions, homeostasis of extracellular PO43– is coordinated between intestinal absorption, renal excretion, as well as its entrance and exit from bone.84 Both parathyroid glands and bone detect increase in extracellular PO43– and they react by increasing the levels of PTH and FGF23, respectively, with the purpose of increasing phosphaturia; however, the intrinsic molecular mechanism sensing the extracellular PO43– is unknown. This is in contrast with what happens with the interactions of calcium or calcimimetics with the RSCa, vitamin D with the VDR and even with FGF23 and its own receptor.33,46,59,75 For many years, attempts have been made to find the PO43– receptor in the parathyroid gland, trying to explain its direct effect, and even postulating the possibility of the existence of a transporter channel.
It is only recently that the importance of the CaSR in sensing the extracellular level of PO43– has been demonstrated. The CaSR is found in the membranes of a variety of cells and belongs to the family of G protein- coupled receptors. It was already known that calcimimetics could overcome the stimulatory effect of increasing PO43– levels on the secretion of PTH in vitro and in vivo.85 Recently, Geng et al.86 used an X-ray crystallography technique to study the three-dimensional structure of the outer domain of the RSCa in active and resting states. It was observed that calcium ions are the main activators of the RSCa but it requires the binding of amino acids in its active form. Likewise, the extracellular domain of the RSCa was also found to have four multivalent anion binding sites occupied by PO43– and SO42– and that PO43– ions kept stable the inactive form of the CaSR, thus promoting PTH secretion. Studies by Centeno et al.87 recently showed in experimental murine models that the increase in PO43– in pathophysiological concentrations of CKD inhibits the activity of the RSCa in an antagonistic, non-competitive way. Thus, these findings finally show us that the RSCa is also a sensor of PO43–, explaining the intrinsic mechanism by which PO43– directly stimulates PTH secretion and providing a mechanism by which elevated concentrations of PO43– can exert direct effects on tissues expressing the RSCa.
In summary, PO43– overload leads to the activation of various direct and indirect mechanisms aiming to maintain its homeostasis.88 The presence of CKD creates at least two vicious cycles in CKD-MBD, the increase in PTH and in FGF23, with the consequent deleterious effects (double "trade-off" that not only affects bone but also the cardiovascular system),1,8,68 and these undesirable effects are the consequence of a failed attempt to normalize mineral metabolism. With the progression of CKD, there is an accumulation of PO43–, and therefore it is important to especially restrict the sources of inorganic PO43– to prevent the development of SHPT; the stimulation of FGF23, the inhibition of calcitriol synthesis and the resulting hypocalcemia, its negative effect on the calcemic response to the action of PTH or the stabilization of the inactive form of the RSCa. PO43– clearly blocks all known counterregulatory mechanisms designed to maintain mineral metabolism homeostasis in healthy situations. In addition, this accumulation of PO43– directly contributes to clear harmful effects mediated by multiple mechanisms that affect the cardiovascular system or the kidney itself1,8,12,89–92 and which seem to explain the very important association of PO43– overload on morbidity and mortality in patients with CKD.
Conflict of interestsJB has received honoraria as a consultant, speaker, or travel support from Amgen, Abbvie, Sanofi, and Vifor-Fresenius-Renal Pharma. PU has received fees as a consultant or speaker from Amgen, Astellas, GSK, Hemotech, Leo-Pharma, Sanofi, and Vifor-Fresenius-Renal Pharma. AT has received consulting fees from Alnylan Pharm. JFNG has received honoraria as a consultant, speaker, or travel support from Abbvie, Amgen, Sanofi-Genzyme, Shire, and Vifor-Pharma. JF has received fees as a consultant from Vifor-Pharma.
Some authors belong to the Renal Research Network (REDINREN RD 16/0009/0022) and RICORS2040 (RD21/0005/0013), Carlos III Health Institute. We also thank Mr. Ricard Pellejero for his invaluable bibliographic help.



