



Hereditary spherocytosis is clinically and genetically heterogeneous disorder and its clinical characteristics are spherocytosis, anaemia, jaundice and splenomegaly. The aetiology is associated to the genes encoding proteins involved in the interaction between the erythrocyte membrane and the lipid bilayer. Causative variants in βI-spectrin (SPTB) gene presenting as mild to moderately severe disease are responsible for approximately 25% cases in the USA and Europe. Among kidney disease, isolated cases of nephrotic syndrome due to membranoproliferative glomerulonephritis and macroscopic haematuria with proteinuria due to IgA nephropathy were previously reported in patients with SPTB deficiency.
ObjectiveSeven patients from the same family with spherocytosis were evaluated to assess the kidney failure presented in all affected adult patients.
MethodsClinical, radiological and laboratory investigations were issued to evaluate the spherocytosis and kidney disease. In selected patients, we also performed genetics testing with next generation sequencing of genes related to hereditary spherocytosis, inherited glomerular disorders and tubulo-interstitial kidney disease.
ResultsAmong the family members with spherocytosis, two adults had end-stage kidney disease and one chronic kidney disease stage 4 with unspecific histopathological findings of interstitial fibrosis/tubular atrophy and glomerulosclerosis. At the time, there were no signs of kidney disease present in four paediatric patients. Novel nonsense variant in SPTB gene (NM_001024858; c.4796G>A; p.Trp1599Ter) was detected in all family members with spherocytosis and was predicted to be disease causing. Furthermore, all adult patients with kidney failure and two paediatric cousins of the index patients were heterozygous for the UMOD gene variant (NM_003361.3:c.552G>C, NP_003352.2:p.Trp184Cys) previously reported in patients with tubulo-interstitial kidney disease. UMOD variant was not present in the index patients.
ConclusionsThe co-occurrence of any two rare inherited disorders is extremely rare, while to our knowledge the co-occurrence of genetically confirmed HS and autosomal dominant tubulo-interstitial kidney disease (ADTKD) has previously not been reported. It is not possibly to evaluate whether the haemolytic crises due to HS are influencing the progression of the UMOD related renal disease, since the UMOD related ADTKD characteristics in general and in here presented family are extremely variable. Nevertheless, the observed kidney disease in the family is warranting the regular nephrological examinations in UMOD positive paediatric patients in the family in order to recognise hyperuricemia and treat it as early as possible. This is emphasising the importance of serum uric acid detection in routine laboratory screening of paediatric patients in order to identify early signs of tubular injury indicating possible ADTKD.
La esferocitosis hereditaria es un trastorno clínica y genéticamente heterogéneo, y sus características clínicas son esferocitosis, anemia, ictericia y esplenomegalia. Su etiología se asocia con los genes que codifican proteínas implicadas en la interacción entre la membrana de los eritrocitos y la bicapa lipídica. Las variantes causativas en el gen de la espectrina βI (SPTB) que se presentan como enfermedad entre leve y moderadamente grave son responsables de aproximadamente el 25% de los casos en EE.UU. y Europa. En el contexto de la enfermedad renal se han notificado casos aislados de síndrome nefrótico debido a glomerulonefritis membranoproliferativa y hematuria macroscópica con proteinuria debido a nefropatía por IgA en pacientes con deficiencia de SPTB.
ObjetivoSe estudió a 7 pacientes pertenecientes a una misma familia con esferocitosis para evaluar la insuficiencia renal presente en todos los pacientes adultos afectados.
MetodologíaSe realizaron investigaciones clínicas, radiológicas y analíticas para evaluar la esferocitosis y la nefropatía. En una selección de pacientes también se realizaron análisis genéticos mediante técnicas de secuenciación de nueva generación de genes relacionados con la esferocitosis hereditaria, los trastornos glomerulares hereditarios y la nefropatía tubulointersticial.
ResultadosEntre los miembros de la familia con esferocitosis 2 adultos presentaban nefropatía en fase terminal y uno nefropatía crónica de fase 4, con hallazgos histopatológicos inespecíficos de fibrosis intersticial/atrofia tubular y glomeruloesclerosis. En ese momento no existían signos de enfermedad renal en 4 pacientes pediátricos. Se detectó una nueva variante sin sentido en el gen SPTB (NM_001024858; c.4796G>A; p.Trp1599Ter) en todos los miembros de la familia con esferocitosis y se predijo que era responsable de la enfermedad. Además, todos los pacientes adultos con insuficiencia renal y 2 pacientes pediátricos primos de los pacientes iniciales eran heterocigóticos para la variante del gen UMOD (NM_003361.3:c.552G>C, NP_003352.2:p.Trp184Cys) notificada previamente en los pacientes con nefropatía tubulointersticial. Los pacientes iniciales no presentaban la variante UMOD.
ConclusionesLa presentación conjunta de cualquiera de estos 2 trastornos hereditarios poco frecuentes es extraordinariamente excepcional, mientras que hasta donde alcanza nuestro conocimiento la presentación conjunta de esferocitosis hereditaria y nefropatía tubulointersticial autosómica dominante (ADTKD) no se había notificado previamente. No es posible evaluar si las crisis hemolíticas debidas a la esferocitosis hereditaria influyen en la progresión de la nefropatía relacionada con el gen UMOD, ya que las características de la ADTKD relacionada con el gen UMOD, en general y en la familia objeto del estudio, son extremadamente variables. Sin embargo, la nefropatía observada en la familia aconseja realizar exploraciones nefrológicas regulares en los pacientes pediátricos positivos para UMOD de la familia, con el objetivo de reconocer la hiperuricemia y tratarla lo antes posible. Esto enfatiza la importancia de la detección del ácido úrico sérico en las pruebas analíticas rutinarias de los pacientes pediátricos para identificar síntomas tempranos de lesiones tubulares indicativas de una posible ADTKD.
Hereditary spherocytosis (HS) is a common type of hereditary haemolytic anaemia. It is clinically and genetically heterogeneous disorder, with spherocytosis, anaemia, jaundice and splenomegaly as typical clinical characteristics.1 Clinical severity is variable with most patients having a well-compensated haemolytic anaemia. Some individuals are asymptomatic, whereas others have severe haemolytic anaemia requiring erythrocyte transfusion. Patients are classified by clinical features as severe, moderate or mild using the criteria establish by Eber SW.2 The assessment should be issued when the patient is in a stable baseline state, as with the undercurrent illness the severity may be overestimated. Children with severe HS are rare (about 5%). They are constantly anaemic and may be transfusion dependent, especially in the first few years of life.
The primary causes of the disease are abnormalities of the red blood cell membrane structural proteins leading to the loss of membrane surface area, erythrocytes deformability and decrease of stability, finally resulting in haemolysis. The aetiology is related to the genes encoding proteins involved in the interaction between the erythrocyte membrane and the lipid bilayer, namely ankyrin (ANK1), α1-spectrin (SPTA1), βI-spectrin (SPTB), band 3 (SLC4A1) and protein 4.2 (EPB42).3 Variants in SPTB gene are causing HS type 2 (MIM#616649) and are inherited in an autosomal dominant manner. They are responsible for approximately 25% of HS cases in the United States of America and Europe presenting with mild to moderately severe disease where transfusion is usually not needed.1,3 Sporadically, large genomic deletions of SPTB gene were associated to psychomotor developmental delay and autism4,5 while SPTB gene substitution was associated to prenatal hydrops fetalis.6 Additionally, variants in SPTB gene were associated to hereditary elliptocytosis (MIM#617948). Among kidney disease, isolated cases of nephrotic syndrome due to membranoproliferative glomerulonephritis7 and macroscopic haematuria with proteinuria due to IgA nephropathy8 were reported in patients with HS. Here we present a three-generation family with seven patients with spherocytosis due to the novel variant in SPTB gene (NM_001024858; c.4796G>A; p.Trp1599Ter). Among the patients in the family, two adults had end-stage kidney disease and one chronic kidney disease (CKD) stage 4 with histopathologicaly unspecific findings of interstitial fibrosis and focal segmental glomerulosclerosis (FSGS) that was later on attributed to tubulo-interstitial kidney disease due to disease causing UMOD variant.
Material and methodsPatientsInitially, two Slovenian paediatric patients with spherocytosis were referred to the paediatric nephrologist at the outpatient clinic of the Department for Nephrology at the University Children's Hospital in Ljubljana, Slovenia, because of the positive family history of CKD in their father, paternal grandmother and uncle. After initial clinical examination, we issued standard laboratory and radiological investigations to evaluate the kidney function, electrolytic disturbances, acid-base status and red blood cell status properties. Additionally, detailed family history regarding renal and haematological diseases was assessed. Later on, all seven affected family members were invited for additional evaluation. Participants consented to the study, which was conducted according to the principles in the Declaration of Helsinki.
Genetic analysisGenomic DNA of all affected family members was isolated from peripheral blood samples using FlexiGene DNA isolation kit (Qiagen, Hilden, Germany). To evaluate the genetic cause of the spherocytosis and CKD in the family a targeted next generation sequencing (NGS) was conducted in both siblings and their affected father and paternal grandmother. We used TruSightOne Sequencing Panel on the MiSeq platform desktop sequencer coupled with MiSeq Reagent kit v3 (all Illumina, USA) followed by on-board primary analysis. A list of rare genetic variants (minor allele frequency <1%) in 5 genes related to HS, 51 genes related to inherited glomerular disorders with FSGS or 4 genes related to autosomal dominant tubulo-interstitial kidney disease (ADTKD) (Supplementary material 1) was filtered with Variant Studio 2.3 software (Illumina, USA). Variants that were identified as possibly causative were confirmed by targeted Sanger sequencing using custom oligonucleotides, BigDye Terminator v3.1 sequencing kit and ABI Genetic Analyser 3500 (both Applied Biosystems, USA) in all family members.
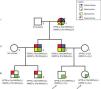
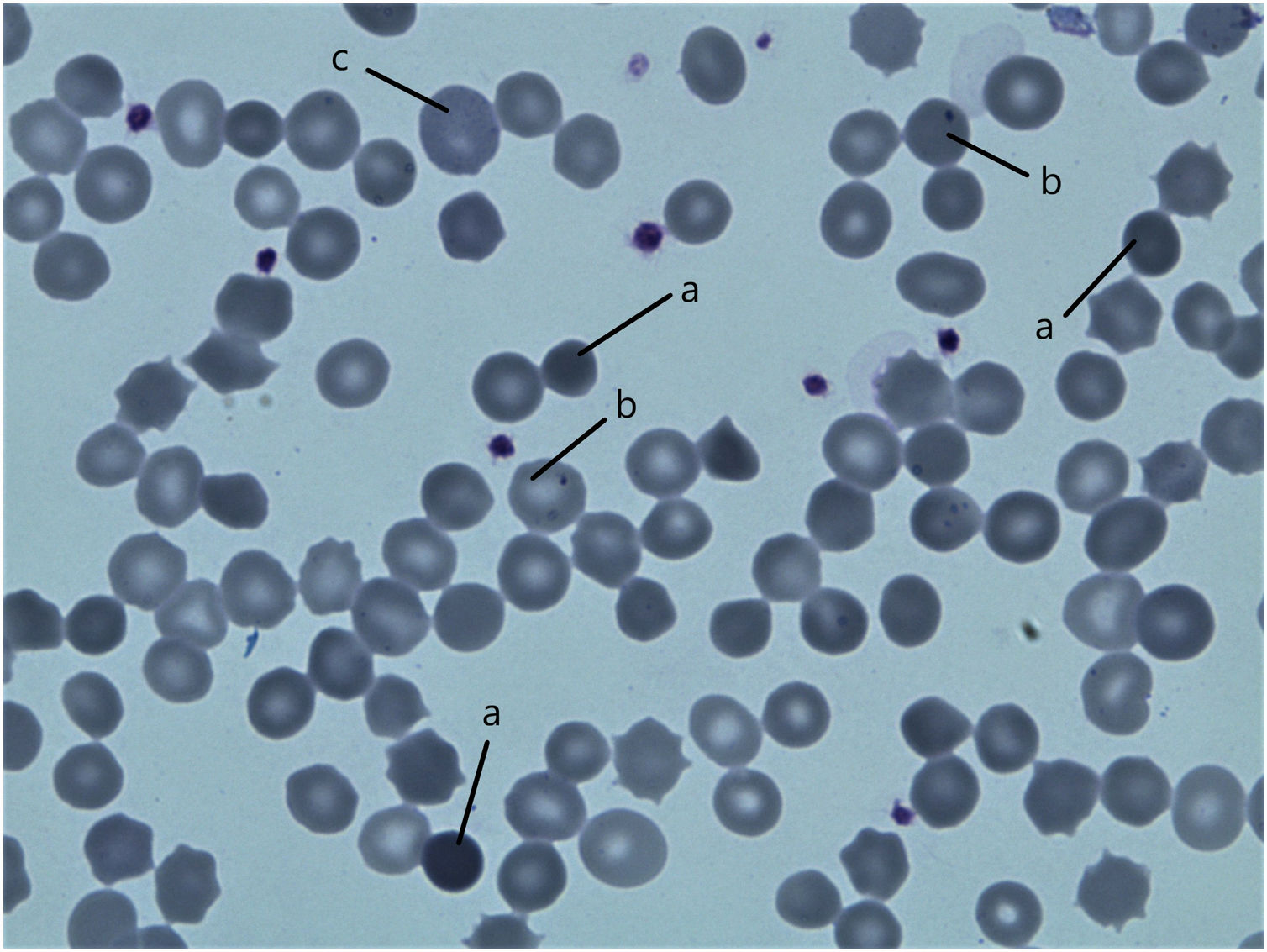
ResultsPatientsFamily pedigree with major clinical manifestations and detected disease-causing variants was summarised in Fig. 1. All family members with spherocytosis had 1–6% of spherocytes in the peripheral blood smear. Additionally, Howell Jolley bodies and occasional basophile punctuations were present (Fig. 2). Mean cell volume (MCV) values were lower than in normal population ranged from 74 to 86fL and median haemoglobin concentrations were 116g/L (95–163). Osmotic fragility test showed increased RBC membrane fragility with tailed sigmoid curve in all patients with HS but patients II-1 and III-4, while RBC band 3 protein fluorescence staining with eosin-5-maleimide (EMA) was significantly decreased in all patients.
Both initially evaluated siblings (subject III-1 and III-2) had normal kidney function and blood pressure, no proteinuria or haematuria, no electrolytic disturbances or acid-base status abnormalities. The ultrasound exam of urinary tract was normal. The older sibling, 16-year-old girl (subject III-1) had so far only one haemolytic crisis that needed transfusion, the younger, 12-year-old boy (subject III-2) had none.
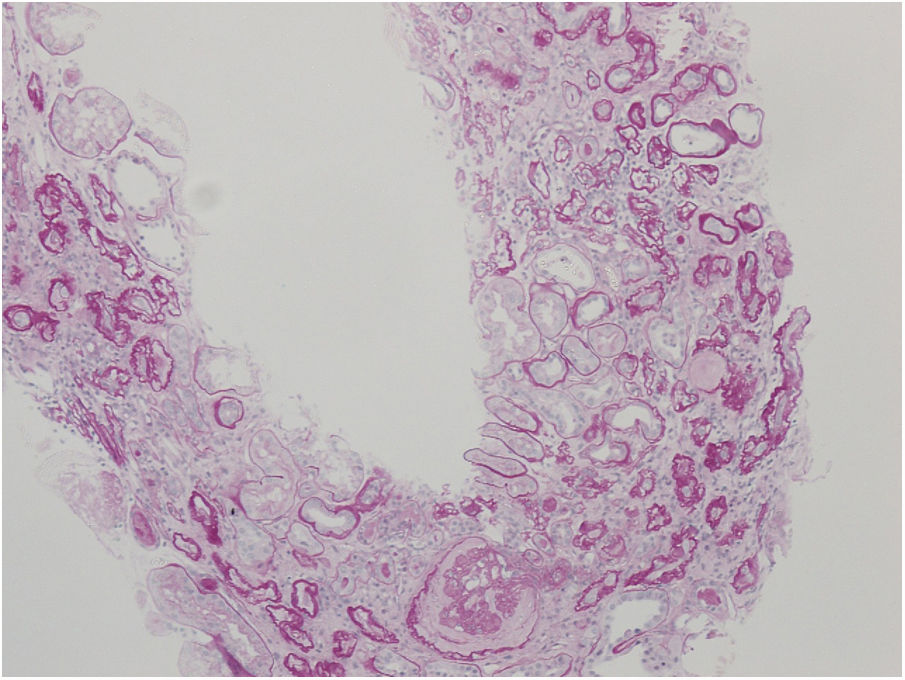
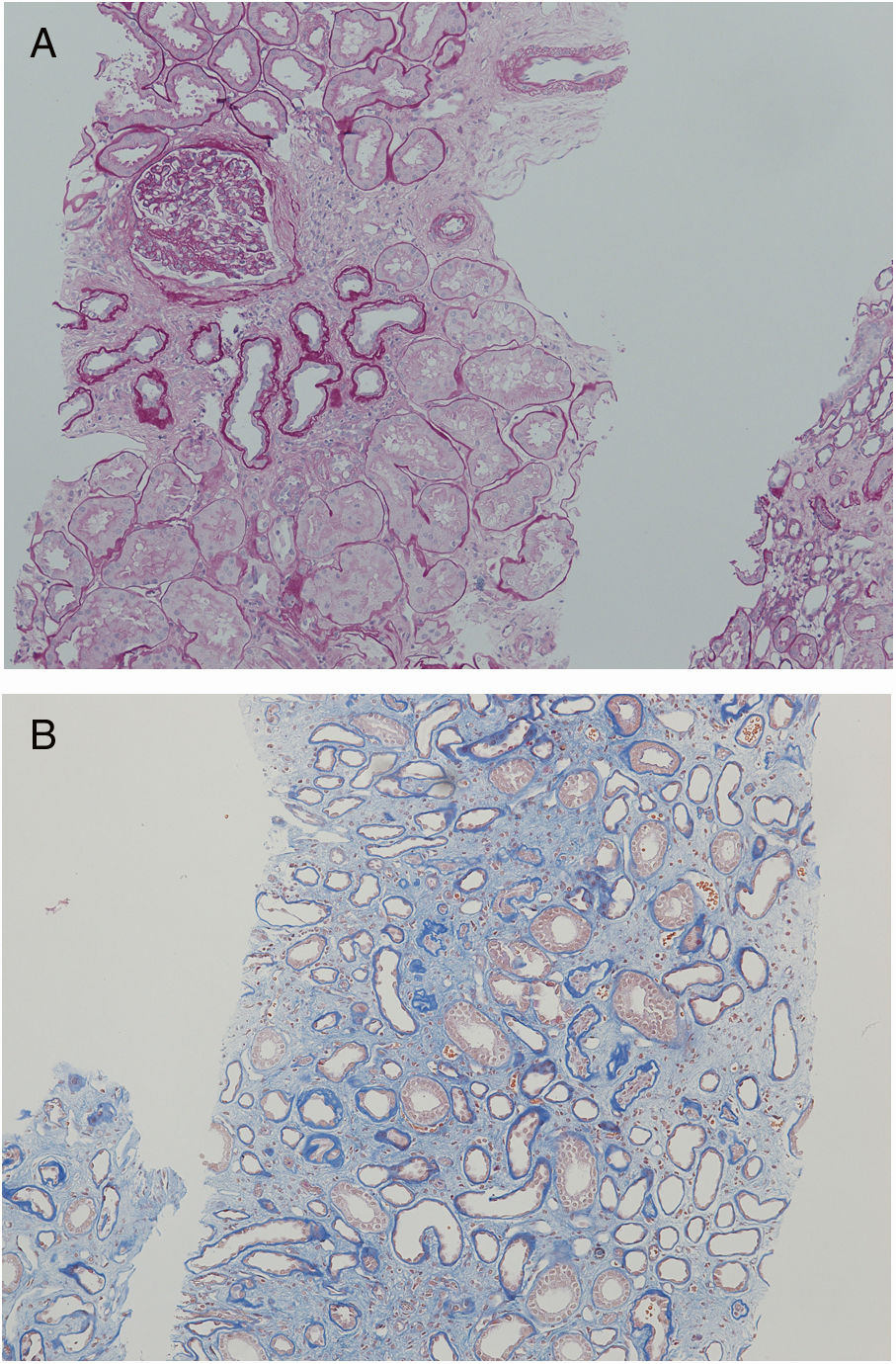
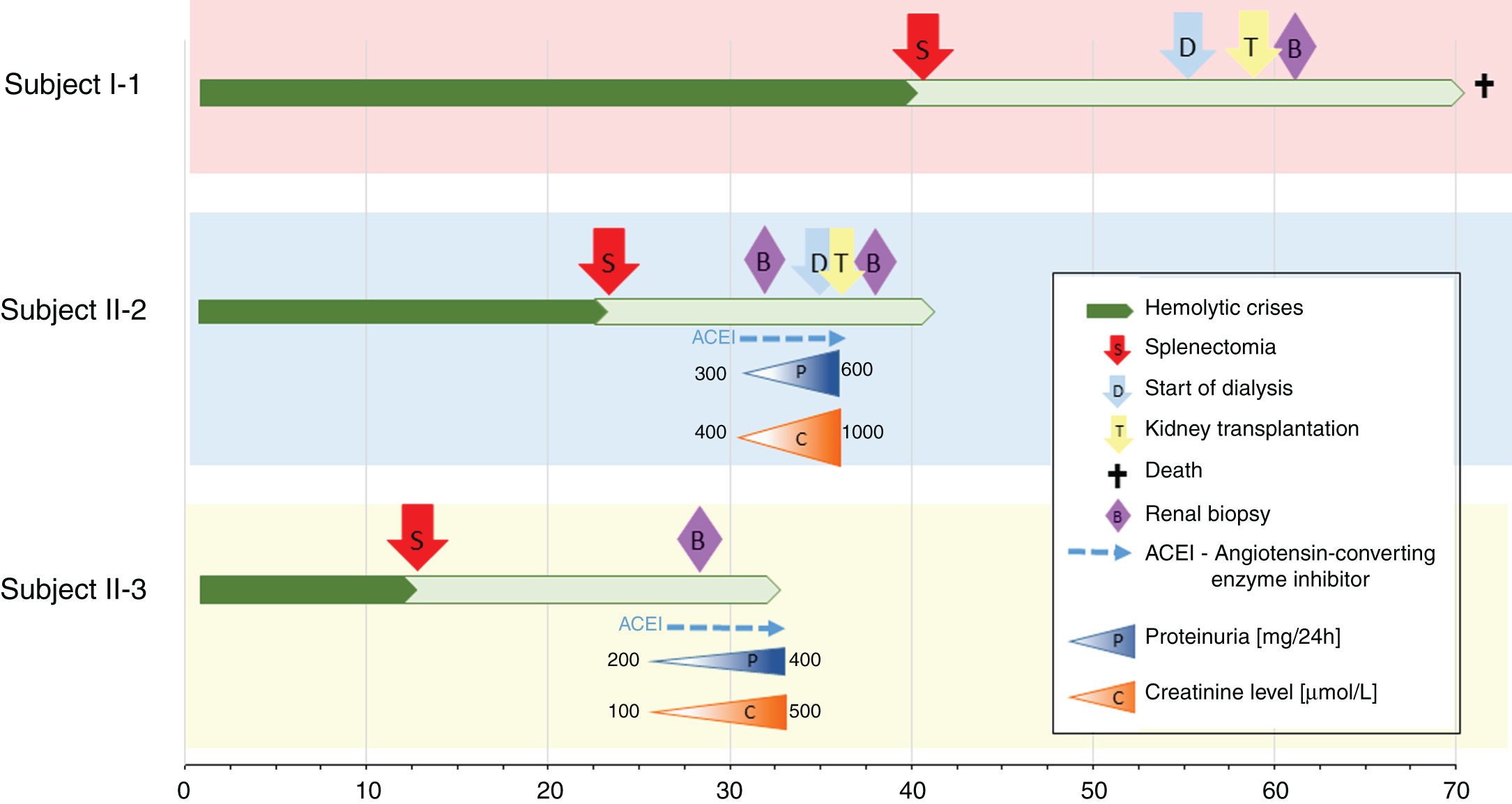
Nephrologist followed the children's 42-year-old father with spherocytosis (subject II-2) due to deterioration of the renal function with hypertension. Splenectomy was performed when he was 22 years old, after one of the many haemolytic crises. After splenectomy haemolytic crises did not repeat. He had hyperuricemia without clinical presentation of gout. When renal biopsy was performed, glomerular filtration rate (GFR) 17ml/min/1.73m2, electrolyte imbalance of potassium and phosphate were described and mild proteinuria (520mg/day). By ultrasonography, his kidney appeared hyper-echogenic and were smaller than expected for an adult male (8cm). Histopathological findings showed rather unspecific changes including diffuse 70% interstitial fibrosis and tubular atrophy with scarce interstitial infiltrate composed of lymphocytes and monocytes, and advanced glomerulosclerosis in 75% of glomeruli (70% global glomerulosclerosis and 5% segmental glomerulosclerosis) (Fig. 3), while no cysts were detected. Electron microscopy revealed normal ultrastructure of glomerular basement membrane with minimal segmental 5% podocyte effacement. Immunofluorescence was unremarkable. He had kidney transplantation three years later, following few months of dialysis. Protocol biopsy one year after transplantation revealed well-functioning kidney graft without signs of rejection, his uric acid was normal without therapy.
The siblings’ paternal grandmother with spherocytosis (subject I-1) had splenectomy when she was about 40 years old. She started with dialysis when she was about 55 years old. She had hyperuricemia that was well controlled by allopurinol and diet. At age of 58, she had kidney transplantation and experienced T-cell mediated rejection 4 months later. She presented with Pneumocystis jirovecii pneumonia and CMV disease resulting in decrease of immunosuppressive therapy and finally lost her graft 3 years after transplantation due to chronic active T-cell and vascular rejection. At the time, she had no follow-up by the nephrologist before the kidney failure, no renal biopsy and there is no available data regarding prior development of her kidney disease. Recently she had passed away due to a stroke in her seventies.
Another family member, 32-year-old uncle of the siblings, namely their father's brother (subject II-3), had splenectomy at age of 12 after several mild haemolytic crises. Later on, the diagnosis of CKD was set. He had gout with many attacks despite of allopurinol therapy and diet. Light microscopy detected 40% interstitial fibrosis and tubular atrophy affecting cortex and medulla with lack of functioning tubules particularly in medulla without any cysts. Accompanied 25% glomerulosclerosis and pericapsular fibrosis of glomeruli indicated secondary involvement due to tubulointerstitial damage (Fig. 4). Immunofluorescence was unremarkable. Additionally, his 2-year- and 4-year-old sons (subjects III-3 and III-4) had spherocytosis with no haemolytic crisis so far and no clinical or echocardiographic signs of kidney disease. Their creatinine clearances were normal, they had no proteinuria, but both had high serum uric acid levels and low fractional excretions of urinary uric acid. Detailed description of CKD development in adult patients over their life span was summarised in Fig. 5.
The analysis revealed that both index patients, their father, paternal grandmother, father's younger brother and his two children were heterozygous for the SPTB variant NM_001024858:c.4796G>A resulting in premature termination codon at the position 1599 (NP_001020029.1:p.Trp1599Ter) (Fig. 1). The variant had so far not been reported in patients with spherocytosis nor in apparently healthy population while in silico tools were predicting it to be pathological (Mutation taster, CADD score 45). According to the ACMG/AMP 2015 guidelines9 the variant was classified as pathological with the following grades: PVS1 (null variant), PM2 (absent from general population), PP1 (co-segregation with the disease in multiple affected family members in gene definitely known to cause the disease), PP3 (multiple lines of computational evidence supporting the deleterious effect). Additionally, we performed NGS based analysis of genes related to inherited glomerular disorders and ADTKD. Father of the index patients, paternal grandmother, father's brother and his two children were heterozygous for the UMOD variant NM_003361.3:c.552G>C resulting in substitution of tryptophan to cysteine at the position 184 (NP_003352.2:p.Trp184Cys) (Fig. 1). This variant has already been reported in patients with ADTKD (HGMD acc. nr. CM118889)10 and was not present in index patients nor in their mother.
DiscussionHS is a common type of hereditary haemolytic anaemia, rarely accompanied by renal manifestations.7 We present a family with seven members presenting with HS due to the novel SPTB variant p.Trp1599Ter suspected to cause βI-spectrin deficiency. In the same family, all three affected adult family members had CKD that was later on attributed to the known UMOD gene variant p.Trp184Cys.
Contrary to the HS in sickle cell disease (SCD), another type of haemolytic anaemia, numerous renal syndromes were described, including increased GFR, acute renal failure, renal tubular disease, chronic renal insufficiency, renal tubular acidosis and others as reviewed in Pham et al.11 Acute kidney injury during SCD pain crisis may be an important risk factor for developing CKD.12 Development of kidney disease in SCD is not fully elucidated. It is associated with oxidative stress caused by the vaso-occlusive episodes, ischaemia-reperfusion injury and the associated chronic inflammatory response, glomerular hyperfiltration and chronic exposure of renal tubular epithelium to high levels of filtered plasma proteins due to proteinuria, as reviewed by Wesson.13 Proximal tubule dysfunction, including tubular proteinuria, is a significant complication in SCD that can eventually lead to CKD. Haemoglobin dimers released from red blood cells upon haemolysis are filtered into the kidney and internalised by megalin/cubilin receptors into proximal tubule cells. Proximal tubule cells are especially sensitive to haem toxicity and tubular dysfunction in SCD is thought to result from prolonged exposure to filtered hemoglobine.14 The mechanism behind the development of kidney disease in HS is not clear and might be related to the mechanism in SCD, since haemolytic crisis are probably the initiators of its development in both cases. Involvement of renal cortex and medulla in the absence of other known causes of medullary tissue remodelling (diabetes, chronic infections, NSAIDS and congenital disorders) suggested haemolysis as one of the potentially pathogenic mechanism of tubular and/or vascular injury leading to interstitial fibrosis and secondary glomerulosclerosis.
In here presented family, we initially speculated that haem toxicity was the cause of the CKD, especially since it had an onset in the adulthood after many haemolytic crises. Nevertheless, to eliminate the less probable existence of additional genetic factors that could contribute to the co-occurrence of the renal disease, we have analysed genes associated to the inherited glomerular disorders and ADTKD. To our surprise, we have identified a variant p.Trp184Cys in UMOD gene in all three adult patients with kidney failure. It was previously reported in 3 members of a Caucasian family with ADTKD, but their clinical presentation was not individually described.15 In general, early onset hyperuricemia is a hallmark of the ADTKD due to UMOD deficiency, where renal survival and clinical characteristic are extremely variable even in patients in the same family. Disease causing UMOD variants are resulting in deficient production of functional uromodulin normally expressed in tubular cells and intracellular deposition of mutant uromodulin. This is leading to accelerated tubular damage and consequently end-stage renal kidney disease.16 Renal biopsy in two adult patients revealed rather unspecific histological changes including interstitial fibrosis, tubular atrophy, glomerulosclerosis and normal podocytes without foot process effacement even in advanced chronic tubulointerstitial changes, which pointed towards tubules/interstitium as primarily affected compartment and is in accordance with the UMOD deficiency. They presented with low proteinuria suggesting FSGS as secondary glomerular changes due to hyperfiltration in remaining overloaded glomeruli. Other risk factors (i.e.: low birth weight, renal asymmetry, obesity, drugs, inherited glomerular disorders) for FSGS development were not present. In addition, two paediatric patients, namely subjects III-3 and III-4, had the same UMOD disease-causing variant. Currently they have no reported haemolytic crises and normal kidney function, but both have high serum uric acid, low fractional excretion of urinary uric acid and low serum uromodulin level confirming deficient production of functional uromodulin.
NGS based genetic testing approach has proved useful as a first-line approach in identification of the genetic aetiology of inherited haemolytic anaemias where several genes can be candidates to be causative.17 Furthermore, in cases with complex clinical manifestations that cannot be directly attributed to one disorder NGS enables analysis of all genes associated to individual manifestation in the same experiment without the need of the strict preselection. This proofed to be extremely valuable in here reported family with co-occurrence of two rare inherited disorders with the clinical onset in different stages of life. Furthermore, the importance of the valid clinical evaluation of the index patients and their family members prior the NGS analysis is well recognised. In here presented family it was crucial since index patients had no kidney disease, but the genetic and clinical evaluation of the family enabled recognition of UMOD related ADTKD in their junior cousins with currently normal kidney function. Consequently, their renal function and serum uric acid level will be closely monitored in order to timely introduce hyperuricemia treatment.18,19 This is emphasising the importance of serum uric acid detection in routine laboratory screening of paediatric patients in order to identify early signs of tubular injury indicating possible ADTKD.
ConclusionsThe co-occurrence of any two rare inherited disorders is extremely rare, while to our knowledge the co-occurrence of genetically confirmed HS and ADTKD has not been previously reported. It is not possibly to evaluate whether the haemolytic crises due to HS are influencing the progression of the UMOD related renal disease, especially, since the UMOD related kidney disease characteristics in general and in here presented family (Fig. 5) are extremely variable. Nevertheless, the observed kidney disease in the family is warranting the regular nephrological examinations in paediatric patients in the family in order to recognise elevated serum uric acid as the first signs of tubular dysfunction, since early onset of hyperuricemia treatment may slow the progression of subsequent kidney disease. The future clinical course in index patients with HS and their two cousins with co-occurrence of HS and UMOD disease-causing variant will highlight the potential role of UMOD related disease as well as HS on kidney function. Clinical data of five patients of different ages with the co-occurrence of two genetic disorders is extremely valuable and my shed a light on the complex pathophysiology of the genetically based co-morbidity.
FundingThe study was supported by the financial support from the Slovenian Research Agency (research core funding P3-0343 and P1-0170).
Conflict of interestNone.
The following are the supplementary data to this article:




