



We describe the case of a young woman who was diagnosed with advanced kidney disease, with an incidental finding of nephrocalcinosis of unknown aetiology, having been found asymptomatic throughout her life. The genetic study by panels of known genes associated with tubulointerstitial disease allowed us to discover autosomal dominant distal renal tubular acidosis associated with a de novo mutation in exon 14 of the SLC4A1 gene, which would have been impossible to diagnose clinically due to the advanced nature of the kidney disease when it was discovered.
Describimos el caso de una mujer joven, que fue diagnosticada de insuficiencia renal avanzada, con un hallazgo casual de una nefrocalcinosis sin una etiología clara, al haberse encontrado asintomática a lo largo de su vida. El estudio genético por paneles de genes conocidos asociados a enfermedad tubulointersticial permitió descubrir una acidosis tubular renal distal autosómica dominante, asociada a una mutación de novo en el exón 14 del gen SLC4A1, que hubiera sido imposible diagnosticar clínicamente por lo avanzado de la enfermedad renal cuando fue descubierta.
Nephrocalcinosis—i.e. a generalised increase of calcium in the kidney—is the result of several factors which contribute to renal damage (nephrolithiasis: elevated urine calcium/oxalate/phosphate, in particular due to serious metabolic defects).1 The main inherited causes of nephrocalcinosis include adenine phosphoribosyltransferase deficiency, cystinuria, Dent's disease, familial hypomagnesaemia-hypercalciuria-nephrocalcinosis, renal tubular acidosis (RTA) and primary hyperoxaluria.2,3
Amongst the latter causes, primary or hereditary distal RTA has gained particular interest in recent years due to de increased understanding of molecular mechanisms that allows to known mutations in the main proteins involved in acid–base transport. RTA is manifested in two hereditary forms: autosomal dominant (AD) and autosomal recessive (AR), which are associated with mutations in the SLC4A1, ATP6V1B1, CA2 and ATP6V0A4 genes. It is characterised by hyperchloraemic metabolic acidosis, which is caused by a failure to excrete hydrogen ions (H+) via the urine. The main symptoms that accompany distal RTA are growth retardation, vomiting/diarrhoea or constipation, poor appetite, polyuria and polydipsia, and nephrocalcinosis. The prognosis for distal RTA is good if diagnosed and treated with bicarbonate/potassium citrate at an early age.4 However, in asymptomatic patients it may be difficult to establish a diagnosis, through just clinical studies/laboratory tests, once advanced renal failure is established, which is also associated with a poor prognosis. In such situations the diagnosis can be obtained through a genetic diagnosis that includes all the known genes associated with the disease.
In this article we describe the case of a young woman with no family history of kidney disease, no prior nephrological examination, who was asymptomatic throughout her life, and, when advanced renal failure was detected, she was diagnosed with renal parenchymal calcification. She underwent a genetic study of all known genes associated with nephrocalcinosis, and was diagnosed with autosomal dominant RTA, identifying a de novo mutation in the SLC4A1 gene as the primary cause of nephrocalcinosis.
Case reportBackground26-Year-old woman with no relevant history or chronic treatment, who was seen by Primary Care Physician because of malaise, cramps and numbness in the hands and feet, and, during the last 2 days, she was unable to open her left eyelid. Lab test was requested which revealed a serum creatinine of 4.6mg/dL (the only previous test recorded available at this centre was 6 years earlier, with creatinine at 1.5mg/dL). Therefore the patient was sent to the emergency room. No family history of kidney disease.
Clinical courseHer blood pressure was 135/67mmHg, heart rate 120bpm and temperature 37.5°C, without relevant finding in the physical exam. The blood test was repeated and confirmed the impaired renal function (creatinine 5.1mg/dL and urea 245mg/dL), together with sodium 124mmol/l; potassium 3.5mmol/l; pH 7.08; bicarbonate 5.9mmol/l; pCO2 20mmHg; corrected calcium ion 0.69mmol/l (normal: 1.13–1.32); magnesium 1mg/dL; WBC 14,060; and a normal CBC and clotting test. Subsequent lab tests showed calcium 7.4mg/dL; phosphorus 5.6mg/dL; iPTH 298pg/ml; vitamin D 18.27ng/ml and uric acid 8.8mg/dL. The immunological exam (immunoglobulins, ANA) was normal. Blood electrophoresis: decreased immunoglobulin. Serological testing for B and C viruses and HIV was negative. In a systematic urine test, pH was 6.0, blood+, and WBC+++. In the 24-h urine protein test, proteinuria was 0.73g, creatinine clearance 12ml/min, uric acid 600mg, urine calcium<4mg/kg and urine oxalate 10mg (normal: 4–31).
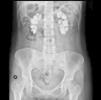
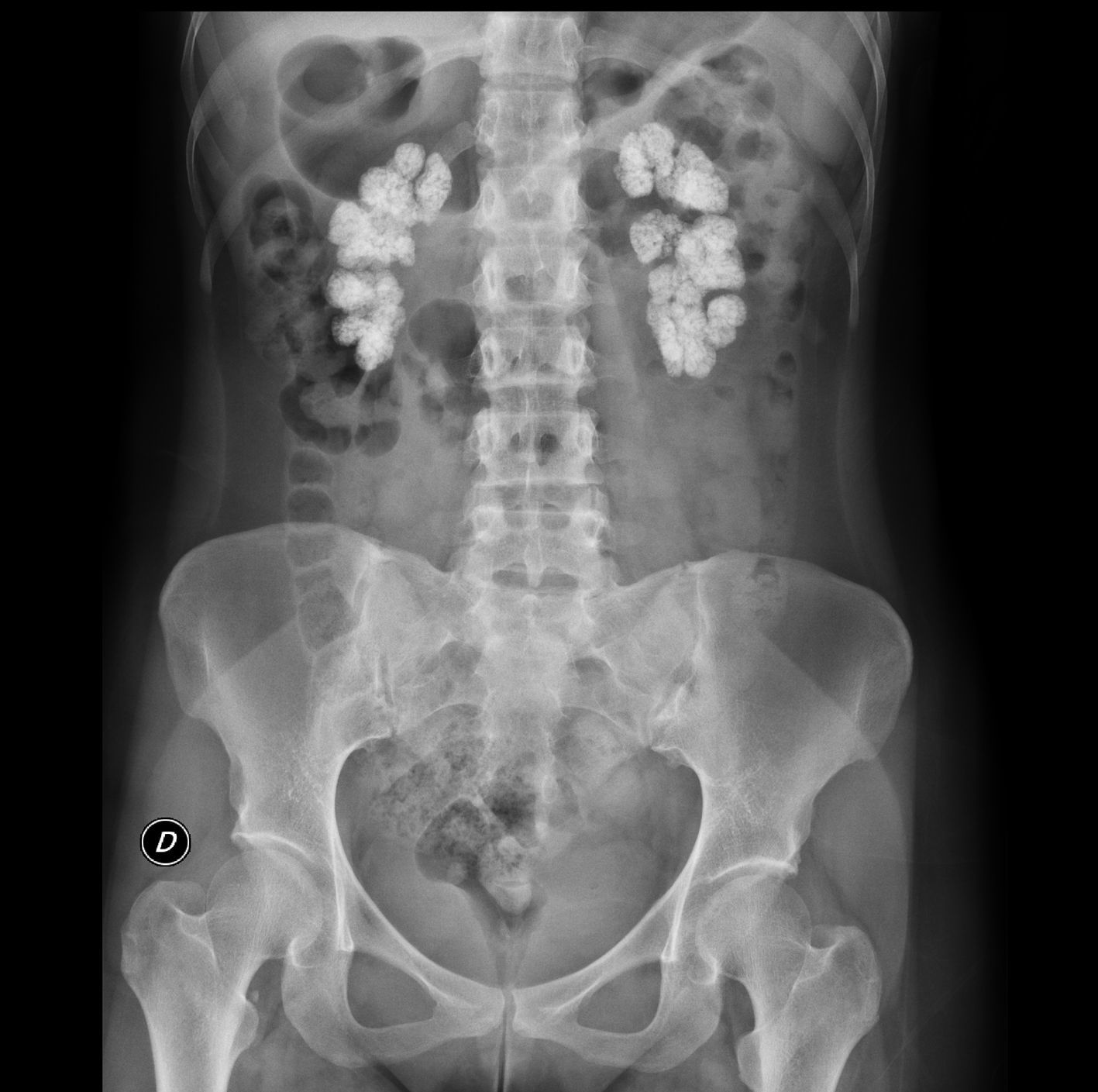
The electrocardiogram showed sinus rhythm with an ST elevation of 1mm in all leads. The chest X-ray was normal. In plain abdominal X-ray, there were extensive bilateral renal calcifications (Fig. 1). After correcting the hypocalcaemia, hypomagnesaemia and metabolic acidosis, the patient was discharged, with no significant incidents during admission. Lab test values at discharge were creatinine 3.6mg/dL; sodium 142mmol/l; potassium 4.4mmol/l; pH 7.43; bicarbonate 24.6mmol/l; calcium 8.6mg/dL; corrected calcium ion 1.16mmol/l; phosphorus 2.9, iPTH 103ng/ml and magnesium 1.9mg/dL. Treatment at discharge was: calcium carbonate 2.5g every 8h; calcitriol 0.5 mcg/day; sodium bicarbonate 1g/day and oral potassium and magnesium supplements.
Approximately 48h after being discharged from the Nephrology Department, she came to emergency room because she was not able to talk or move her tongue. In addition, the patient reported that the previous night she had trouble controlling the movements of her right hand. She also reported trouble swallowing and severe asthenia. She had no fever or headache. The physical exam showed blood pressure 102/72mmHg, heart rate 75bpm, baseline oxygen saturation 98%, and all else was normal. The blood test showed creatinine 3.6mg/dL; sodium 141mmol/l; potassium 4.4mmol/l; pH 7.43; bicarbonate 24.6mmol/l; calcium 8.6mg/dL; corrected calcium ion 1.16mmol/l; and magnesium 1.9mg/dL. The patient was assessed in Neurology and was admitted for examination. In Neurology, an edrophonium test was negative, brain CT without contrast with no significant findings, EEG (abundant outbreaks of paroxysmal activity [theta brain waves] in the left temporal region, with spread to the rest of the hemisphere, as well as the homologous contralateral region, forming some bilateral paroxysmal outbreaks of acute waves). Treatment was started with levetiracetam 250mg/12h, which was insufficient to control the crisis. The patient developed focal status epilepticus with no response to diazepam (up to 10mg) or valproic acid boluses, and so she had to be transferred to the ICU. The seizures were controlled with infusion of valproic acid infusion (1g/24h); since then, the patient has been asymptomatic. In the Neurology ward, she was given oral treatment and remained asymptomatic. She was discharged on oral valproic acid 500mg/12h.
Genetic testGiven the absence of a family history of kidney disease, the current situation of advanced renal failure and the diagnostic difficulties for specific tests to clarify the primary process that led to the development of nephrocalcinosis, it was decided to conduct a genetic test. An analysis of all known genes associated with tubulointerstitial disease (http://nefrochus.villaweb.es/en/) was requested, including an analysis of gene variants found in known genes associated with forms of nephrocalcinosis (“NefroCHUS”, Santiago de Compostela). The test showed that the patient (II: 1) was carrying a missense mutation in exon 14 of gene SLC4A1, associated with autosomal dominant RTA. This mutation (c.1766G>A, Fig. 2A) involves the substitution of a basic amino acid (p.R589H), arginine (Arg, R), for another amino acid, histidine (His, H), which has not been found in our cohort of normal chromosomes, in the international project 1000 genomes, or in the dbSNP137 data base, although it has been described in the literature,5,6 with genomic code rs121912744, catalogued as CLINSIG=pathogenic and associated with families with autosomal dominant distal RTA. The functional testing done, with various in silico prediction tools, assesses gene variant pathogenicity criteria (SIFT: deleterious; polyphen2: deleterious, LRT: deleterious; MutationTaster: disease_causing_automatic; mutation assessor: medium; FATHMM: tolerated; RadialSVM: deleterious and LR: deleterious).
Cosegregation study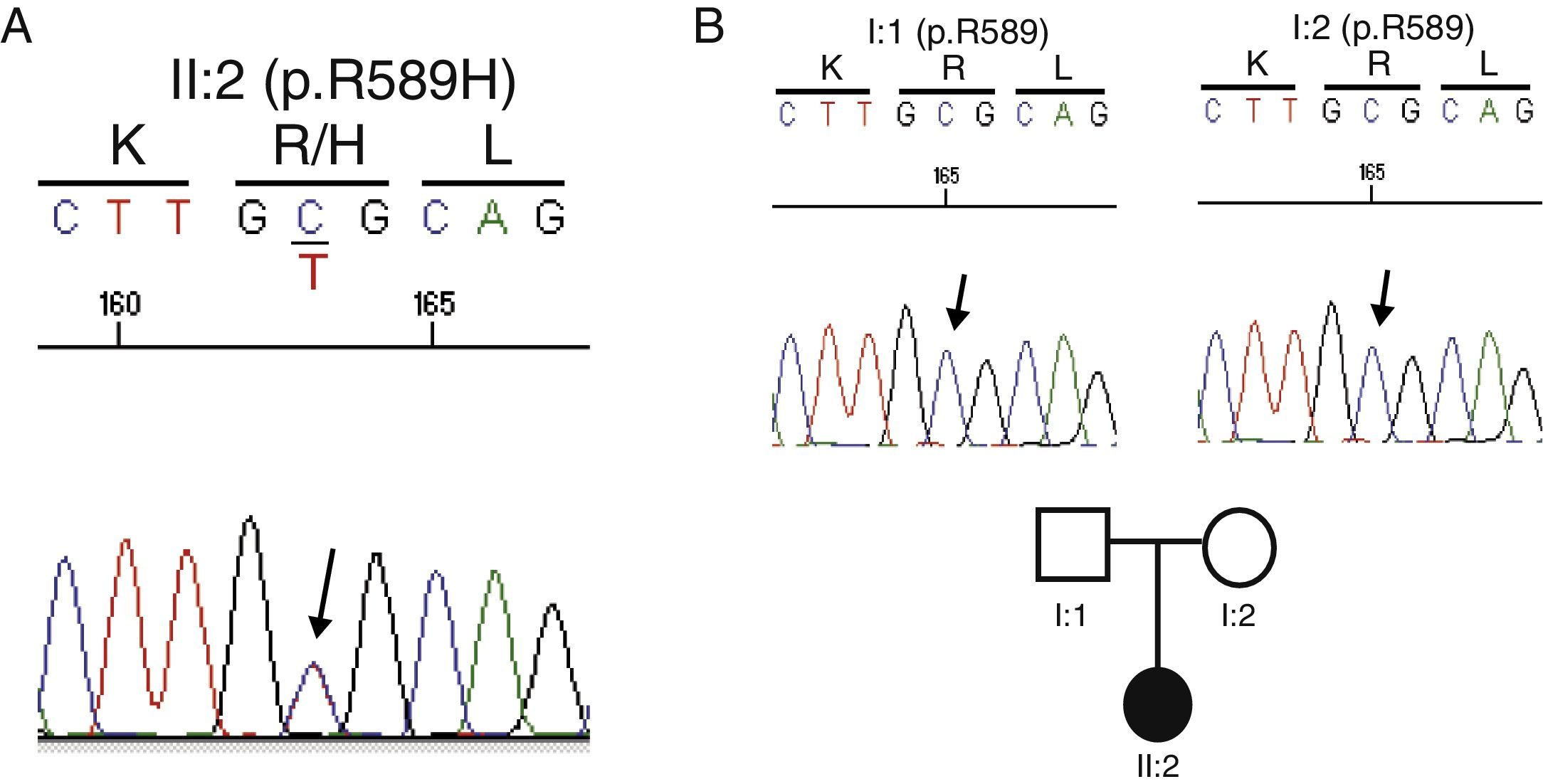
Considering a possible kidney transplant from a living donor, a cosegregation study was performed in both progenitors (I:1 and I:2). Not carrying the R589H mutation indicates that the mutation is de novo or spontaneous, and thus they may be candidates for donation (Fig. 2B).
DiscussionGenetic testing with panels for all known genes associated with tubulointerstitial nephropathy has allowed us to determine that the primary cause of nephrocalcinosis in our patient was due to distal renal tubular acidosis caused by a de novo missense mutation in the SLC4A1 gene previously described in the literature.5,6
In the outpatient clinic, distal RTA can be diagnosed by determining plasma creatinine and fractional sodium, potassium and chlorine clearance, urine calcium and urine citric acid, and in cases of doubt, tests for tubular acidification with NH4Cl may be performed. However, in asymptomatic patients with no family history, this condition may go unnoticed. Once it develops advanced renal failure, functional tests may not be helpful for an accurate diagnosis of the cause of nephrocalcinosis, as in our case, where multifactorial metabolic acidosis and renal tubulopathy were due to the damage established. In fact, in our patient the subsequent onset of seizures led us to consider familial hypomagnesaemia hypercalciuria-nephrocalcinosis as a possible cause of the nephrocalcinosis, which the genetic test eventually ruled out.
This study highlights the importance of genetic testing, because it allowed us to find the primary cause associated with the cause of nephrocalcinosis. We recognise that conducting diagnostic testing involves the following advantages: (1) If there had been a chance for an early diagnosis, the advanced kidney failure could have been prevented in our patient, since early treatment could have been administered. (2) In the case of renal replacement therapy, we could be more confident that the disease would not relapse in the renal graft if we knew the exact cause of the nephrocalcinosis. (3) We could also pre-screen progenitors safely as possible donors, by ruling them out if they carried a mutation. In conclusion, we report the case of a patient with a mutation in the SLC4A1 gene associated with autosomal dominant distal renal tubular acidosis, which caused nephrocalcinosis, and which had gone unnoticed until becoming a clinically advanced stage of renal failure. Without the genetic test that included all genes associated with tubulointerstitial disease, it would have been impossible to make an accurate diagnosis of the main cause of the nephrocalcinosis.
Conflicts of interestThere are no conflicts of interest or funding.
Please cite this article as: Heras Benito M, Garcia-Gonzalez MA, Valdenebro Recio M, Molina Ordás Á, Callejas Martínez R, Rodríguez Gómez MA, et al. Necesidad de estudio genético para el diagnóstico de algunos casos de acidosis tubular renal distal. Nefrologia. 2016;36:552–555.



