







Mitochondrial diseases, taking into account those that affect the processes of the respiratory chain (RC) and mitochondrial oxidative phosphorylation system (OXPHOS), make up a relatively frequent group within rare diseases that usually have multisystem involvement, a very variable phenotypic expression and a complex genetic base. Renal involvement is uncommon, with the tubule being the most affected, specifically its proximal portion, developing into full Toni-Debré-Fanconi syndrome in the most serious cases. However, in some cases the glomerulus is involved, fundamentally in focal segmental glomerulosclerosis form (FSGS), expressed by proteinuria and renal failure. It is important that the Nephrologist keeps in mind the possibility of a mitochondrial disease in patients with this type of renal involvement that present clinical data with these characteristics, especially diabetes mellitus and deafness. In cases with FSGS, a correct diagnosis will avoid the inappropriate use of immunosuppressive medication. Specific treatments do not exist for the majority of mitochondrial diseases, but it is likely that the intense research that currently exists for these diseases will eventually produce effective treatment possibilities.
Las enfermedades mitocondriales, considerando aquellas que afectan a los procesos de la cadena respiratoria (CR) y fosforilación oxidativa mitocondrial (OXPHOS), constituyen un grupo relativamente frecuente dentro de las enfermedades raras que habitualmente tienen afectación multisistémica, una expresión fenotípica muy variable y una base genética compleja. La afectación renal es poco común, siendo el túbulo, y más concretamente su porción proximal, el principal afectado, desarrollándose un síndrome de Toni-Debré-Fanconi completo en las formas más graves. No obstante, en algunos casos existe afectación glomerular, fundamentalmente en forma de glomeruloesclerosis segmentaria y focal (GESF), manifestada por proteinuria e insuficiencia renal. Es importante que el nefrólogo tenga presente la posibilidad de una enfermedad mitocondrial en pacientes con esta forma de afectación renal que presenten datos clínicos acompañantes característicos, sobre todo diabetes mellitus y sordera. En los casos con GEFS, un diagnóstico correcto evitará el uso inapropiado de medicación inmunosupresora. No existen tratamientos específicos para la mayoría de las enfermedades mitocondriales, pero es probable que la intensa investigación actualmente existente sobre estas patologías lleve finalmente a posibilidades terapéuticas eficaces.
INTRODUCTION
Human mitochondrial deoxyribonucleic acid (mtDNA) is a double-stranded circular molecule that contains 37 genes (Figure 1). Of these genes, 24 encode mitochondrial translation machinery proteins, 22 transfer ribonucleic acids (tRNAs), one for each amino acid except leucine and serine, which contain 2 tRNAs and 2 ribosomal ribonucleic acids (rRNA). The remaining genes encode 13 polypeptides that are part of the enzyme complexes of the respiratory chain (RC). Genes MTND1 – MTND6 and MTND4L are part of complex I (NADH-coenzyme Q oxidoreductase), the MTCYB gene, cytochrome b of complex III (ubiquinol-cytochrome C reductase), genes MTCO1, MTCO2 and MTCO3, of complex IV (cytochrome oxidase). Finally, genes MTATP6 and MTATP8 of Complex V (ATP synthase). Complex II (succinate dehydrogenase) is exclusively formed by subunits encoded in nuclear DNA (nDNA). Most of the assembly and structural proteins of the CR complexes, such as those involved in mtDNA maintenance processes or biogenesis, are encoded by nuclear genes and imported into mitochondria1. Therefore, we can deduce that mitochondrial disorders may be due to sporadic or inherited mutations in both mtDNA and nDNA. The form of inheritance will depend on the location of the mutation, as discussed below.
Kidney disorders described as mitochondrial diseases include a wide spectrum whose most frequent target are tubular cells, due to the high energy expenditure required for ion exchange in these cells, which becomes apparent as proximal tubulopathy, distal tubulopathy or renal tubular acidosis. However, there are ever more frequent cases described in the literature of glomerular involvement in the form of focal segmental glomerulosclerosis (FSGS)2-20 and other less frequent glomerulopathies such as minimal change disease13,14, IgA11 nephropathy or extracapillary glomerulonephritis21. Cystic renal diseases have also been described12,22,23.
MITOCHONDRIAL DISEASES DUE TO mtDNA MUTATIONS
Mitochondria are inherited exclusively through the maternal line, so that mutations in mitochondrial genome will be transmitted from mother to child without paternal contribution. Unlike nDNA, mtDNA has thousands of copies within a cell and mutated DNA can coexist with normal DNA; this state is known as heteroplasmy. Clinical manifestations of mitochondrial disease will occur when the mutated DNA exceeds a limit determined by the energy requirements of each cell, causing cell dysfunction24. For these reasons, mitochondrial diseases have a wide variability in phenotype expression even within the same family.
Mutations that occur in the mitochondrial genome can be point mutations, small and large deletions and duplications. Since the mitochondrial genome is very susceptible to mutation, it has a large number of neutral or adaptive gene variants, known as polymorphisms. Other mutations, a good portion of them heteroplasmic, are deleterious, especially those occurring in the 13 proteins encoded in the mitochondrial genome or rRNA and tRNA. Although most mitochondrial diseases caused by mutations in mtDNA are maternally inherited, some are sporadic, because they occur during oogenesis or early embryogenesis, the most well-known are large (unique) simple mtDNA deletions.
Maternally inherited diabetes and deafness (MIDD)
Maternally inherited diabetes and deafness is one of the most common mitochondrial diseases. As its name indicates, patients have non-insulin dependent diabetes mellitus (NIDDM) produced by a decrease in insulin release from beta pancreatic cells as well as a slowly progressive sensorineural hearing loss. Other accompanying clinical manifestations are cardiomyopathies (hypertrophic or dilated), cardiac conduction disorders in the form of blocks or arrhythmias10, proximal myopathy or fundus alterations, such as pigmented maculopathy, which on rare occasions can decrease visual acuity. About 80% of patients have sensorineural hearing loss or diabetes at the time of diagnosis2-14,25. The most commonly found mtDNA mutation in this disease is a point mutation (m.3243A>G), in the MT-TL1 [tRNALeu(UUR)] gene, leading to inadequate translation of mitochondrial proteins that form CR subunits. Regarding renal involvement, cases of interstitial nephritis12,26, polycystic kidney12 and FSGS have been described2-14,25.
The association between FSGS and mitochondrial disease is not yet well known in nephrology. Early diagnosis of this form of renal involvement is particularly important to prevent patient exposure to ineffective and often toxic immunosuppressive therapies. In addition to knowledge of the disorder, it is essential, for early diagnosis, to have the patient’s complete clinical history and to carry out a physical examination to detect characteristic clinical signs of these disorders, which we summarise in Table 1. In this form of FSGS, the most frequent presentation is non-nephrotic proteinuria with poor response to antiproteinuric treatment, which slowly progresses to end-stage renal disease (ESRD). Approximately 50% of patients will develop ESRD in the first ten years after diagnosis. Because of the association of deafness and renal disease, these patients can be misdiagnosed with Alport disease. The absence of haematuria in this form of FSGS is important for differential diagnosis, since haematuria is a constant finding in Alport disease.
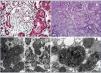
On histological exam, neither the FSGS lesions observed by optical microscopy nor the absence of immune complex deposition by immunofluorescence (IF) make it possible to suspect a secondary form of mitochondrial disease, since no subtype is predominant. However, when there is sufficient material available for electron microscopy, it is frequently possible to see abnormal mitochondria of different shapes and cristae with distortion of the inner membrane and ridges (Figure 2). Accumulations of these mitochondria are located in the tubular cells and podocytes, but are absent in capillary mesangial and endothelial cells. In addition, podocytes are frequently binucleate and undergo focal pedicel fusion. Another striking frequently observed disorder is hyalinosis of smooth muscle cells of afferent arterioles and small arteries. Indeed, according to microscopic findings two pathophysiologic mechanisms for FSGS have been hypothesised in this disease, which are still to be elucidated. On the one hand, the authors who found intense arteriolar hyalinosis in renal biopsies consider that glomerular sclerosis could be due to an alteration in the self-regulation mechanism that would lead to excessive glomerular pressure and consequent hyperfiltration7,9. As in MELAS (Mitochondrial encephalomyopathy, Lactic Acidosis and Stroke-like Episodes), these authors believe that the main damage occurs at the level of the vascular smooth muscle, where abnormal mitochondria have also been found27. However, the authors who found megamitochondria at podocyte level8,10-13,25 speculate that glomerular injury is produced by direct podocyte damage secondary to mitochondrial dysfunction in these cells.
Below we summarise three clinical cases diagnosed between the years 2012-2014 in the Nephrology Service of the 12 de Octubre Hospital, including, the most frequent clinical symptoms of this disease and, confounding diagnostic facts that may arise.
First case14: a 38-year-old male patient, with a history of NIDDM and hypertension (HTA) of three years of evolution, hypertrophic cardiomyopathy, and sensorineural hearing loss from youth. The patient is referred to the nephrology clinic due to non-nephrotic proteinuria and chronic renal failure with creatinine of 2.4mg/dl, and suspected Alport syndrome. Family history: his mother is diabetic, with hearing loss and chronic renal failure of unknown aetiology. After a thorough study, including urine sediment without microhaematuria, and negative autoimmunity and serological tests for hepatitis B, hepatitis C and human immunodeficiency virus, a kidney biopsy is performed. This shows eight glomeruli, of which three are sclerotic, two present focal segmental sclerosis lesions with hyalinosis, two marked capsular fibrosis and the others slight mesangial hypercellularity. In addition, there is intense arteriolar subendothelial hyalinosis with marked lumen reduction. IF is negative and ultrastructurally megamitochondrias were observed in the podocytes (Figure 2 A and Figure 2 C). Due to the association of NIDDM, deafness, kidney disease and hypertrophic cardiomyopathy in a patient who also has a sediment without haematuria, mitochondrial disease is suspected and confirmed after a genetic study.
Second case14: 19-year-old woman with a history of high blood pressure, sensorineural hearing loss from youth, non-autoimmune hypothyroidism and pigmented maculopathy of the fundus, with no relevant family history. The patient is referred to nephrology due to non-nephrotic proteinuria. Studies performed included urinary sediment without microhaematuria, negative autoimmunity and serology tests. At that time, a kidney biopsy puncture was performed which yielded insufficient material. And was therefore repeated 5 years later. This new biopsy has eight optically normal glomeruli, IF with no relevant findings and an electron microscopy image not compatible with Alport disease, with megamitochondria showing ridge distortion and an unstructured inner matrix (Figure 2 B and Figure 2 D). In her third pregnancy (during the first two she suffered early miscarriages) the patient develops diabetes and massive nephrotic syndrome. With a probable diagnosis of FSGS decompensated by pregnancy, treatment with steroids and tacrolimus is initiated, in spite of which adequate control of nephrotic syndrome is not achieved and impaired renal function is evident. Finally, it was decided to terminate the pregnancy due to foetal distress and risk for the mother’s life. Proteinuria returned to non-nephrotic levels and renal function to normal, so immunosuppression was suspended. Once again, in view of the association of diabetes, deafness, steroid-resistant nephrotic syndrome (SRNS) and pigmented maculopathy of the fundus, we requested a genetic study for mitochondrial disease, to confirm our clinical suspicion.
Third case: a 66-year-old woman, diabetic from 34 years of age, currently insulin-dependent with sensorineural hearing loss and significant family burden of type 2 diabetes mellitus on the maternal side. She was referred to the nephrology clinic due to non-nephrotic proteinuria with normal renal function. Because the patient does not have diabetic retinopathy of the fundus, but does have diabetes, deafness and proteinuria with a family history on the maternal side, we requested a genetic study for mitochondrial disease, which, once more confirmed our clinical suspicions. No renal biopsy was performed.
In all three cases, the symptoms were similar: diabetic patient, with hearing loss and renal involvement, non-nephrotic proteinuria and sediment without microhaematuria. Genetic studies showed the same mutation in mtDNA (m.3243A>G in the tRNALeu(UUR)), with very different levels of DNA heteroplasmy in peripheral blood (79% first case, 37% second case and 17% third) detected by direct Sanger sequencing of the MTTL1 gene and quantified by polymerase chain reaction and restriction analysis (PCR-RFLP) of a 1000 DNA chip in an Agilent Technologies 2100 Bioanalyzer, which confirmed diagnosis of MIDD. This variability may be due to a high index of mitotic replacement in white blood cells (WBC) and mitotic segregation of mtDNA. Post-mitotic tissues would be more reliable for these studies. Musculoskeletal system or slowly mitosis cells, such as urinary sediment cells. In all three cases, we began treatment with renin-angiotensin system blockers with very little response as far as proteinuria reduction. The first case, after five years of follow-up, continues with creatinine levels of 2.4mg/dL and non-nephrotic proteinuria. The second case, after sixteen years of evolution, has a normal renal function with non-nephrotic proteinuria. The third case, under follow-up since 2011, has normal renal function and non-nephrotic proteinuria.
In Table 2 we summarise the published clinical series of cases of MIDD and FSGS. Of the 135 patients included in the published papers, 106 (78.5%) had MIDD. Of these 135 patients, in 9 (4) there was no description of presence or absence of hearing loss, but 120 of the remaining 126, did suffer from hearing loss (95.2%). Regarding renal involvement, only 51 patients had proteinuria, and in 4 the degree of this was not specified. Of the remaining 47 patients, 40 (85.1%) had non-nephrotic proteinuria, and 7 (14.9%), nephrotic proteinuria. Only three patients presented a full nephrotic syndrome. In the data obtained from 83 patients, the first symptom of the disease was sensorineural hearing loss in 49 patients (59%), diabetes in 25 (30.1%) and proteinuria in 9 (10.8%). 42.3% (22/52) of those who showed renal involvement ended in dialysis during follow-up. Of the 11 patients reported to have received renal transplant (3,7,9,12), as was to be expected, none had disease relapse in the graft. However, none of the above publications were designed to describe their evolution.
Mitochondrial Encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS)
MELAS syndrome is an encephalomyopathy with lactic acidosis and stroke-like episodes with seizures and dementia. A number of different mutations have been associated with this syndrome (www.mitomap.org), although in 80% of cases it is due to the aforementioned mutation (m.3243A> G in tRNALeu(UUR))28. As with MIDD syndrome, the most characteristic renal disorder is FSGS29, although tubular pathologies and interstitial nephritis have also been described.
Kearns-Sayre
Syndrome characterised by chronic progressive external ophthalmoplegia, ptosis, abnormal heart rhythms, diabetes, deafness, pigmented retinopathy and cerebellar anomalies, that, at times, are accompanied by renal involvement, especially tubular pathologies28,30, although glomerular disorders have also been described30. This mitochondrial disease is usually caused by large deletions at mtDNA levels (Table 3). Martín-Hernández et al. published a series of 42 patients with mitochondrial disease, 21 of whom had renal involvement. Of these, 6 showed well-defined renal disease: 3 Toni-Debré-Fanconi syndromes within a Pearson syndrome, one patient a Kearns-Sayre syndrome and the last patient presented this syndrome secondary to a mutation in the BCS1L gene. In the remaining case, the patient had a nephrotic syndrome secondary to FSGS due to a deficiency of CR complex III44.
Pearson
Syndrome that appears in childhood and typically takes the form of sideroblastic anaemia, exocrine pancreatic failure and renal involvement similar to that seen in Kearns-Sayre syndrome19. Large deletions of mtDNA as causes of this disease have also been reported.
MITOCHONDRIAL DISEASES DUE TO nDNA MUTATIONS
Contrary to mtDNA mutations, mutations in nDNA exhibit Mendelian inheritance, although autosomal dominant and X-linked mutations are known, most are autosomal recessive and therefore, patients’ siblings, and not their parents, may present the disease.
Coenzyme Q10 biosynthesis
Primary defects of coenzyme Q, intermediary redox substrate of complex I, II and III of the CR, are associated with a decrease in mitochondrial oxidative phosphorylation (OXPHOS) that primarily affects the kidneys with little extrarenal expression. The mechanism by which these mutations affect the kidney, specifically the podocyte but not tubular cells, and not other more strongly OXPHOS dependent organs, is still unknown. Six cases have been described with COQ221,45,46 gene mutations, suffering from SNCR and progressive encephalopathy; 11 more cases with COQ6 gene mutations also showing SNCR associated with deafness and seizures47; and 2 more cases, one with a PDSS2 mutation suffering from SNCR and encefalopathy48, and one with a COQ9 mutation suffering from encephalopathy and renal tubulopathy49 (Table 4). The most important characteristic of this set of mutations is that they cause the only mitochondrial diseases with good response to treatment with supplemental oral coenzyme Q. Regarding renal involvement, proteinuria improves with early diagnosis and treatment when there is no chronic renal damage. Some published cases failed to demonstrate these benefits because treatment was initiated when renal damage was advanced50,51. However, Montini et al.52 described a 12-month-old girl with a COQ2 gene mutation who showed initial symptoms of a full nephrotic syndrome and impaired renal function. After immediately starting supplementary treatment (coenzyme Q10 30mg/kg/day) she showed progressive recovery, achieving normal renal function and non-nephrotic proteinuria, which continue after 50 months follow-up52.
Translation of mitochondrial proteins
The aminoacyl tRNA synthetases required for mitochondrial tRNA aminoacylation, are encoded in the nuclear genome. Of the 20 tRNA synthetases, only one has been associated with renal disease, serilARNt synthetase. SARS2 gene mutations encoded by this aminoacyl synthetase, cause a syndrome known as HUPRA (hyperuricemia, pulmonary hypertension, renal failure and alkalosis) with renal failure and distal renal tubular dysfunction (Table 4)53.
The mitochondrial genome encodes two rRNA molecules; however, all ribosomal subunit proteins are encoded in the nuclear genome, so that mutations at that level cause mitochondriopathies. Mutations in MRPS22 ribosomal protein have been associated with renal tubular disease in a patient with cardiomyopathy and hypotonia (Table 4).
Assembly and operation of the respiratory chain
CR defects due to mutations in different genes encoded in the nuclear genome lead to decreased complex activity, mainly, of complexes III and IV. Mutations in BCS1L, the mitochondrial assembly factor necessary for the assembly of complex III54, or in SURF1 gene encoding a protein involved in the biogenesis of complex IV, or mutations in the COX10 gene, the assembly factor for this last complex, have been described in children with renal tubular acidosis proximal tubulopathy and other systemic disorders. In addition, mutations in TMEM70 gene encoding a protein of the baseline membrane for the function of V complex were associated with hypoplastic kidneys in 5 of 25 neonates also suffering from hypotonia, cardiomyopathy and lactic acidosis (Table 4).
Posttranslational processing of mitochondrial proteins and mtDNA depletion syndromes
Defects in post-translational processing of mitochondrial proteins as well as mtDNA depletion syndromes have been associated with renal disease in rare cases, causing tubular atrophy with interstitial fibrosis and chronic renal failure and proximal tubular dysfunction, respectively (Table 3)44.
DIAGNOSTIC ORIENTATION
Knowledge of the association between mitochondrial disease and renal disease is very important, especially in cases of nephrotic syndrome or FSGS, in which diagnosis of mitochondrial disease prevents exposure of patients to immunosuppressive therapy, also in those cases in which the mutation affects biosynthesis of coenzyme Q10, where early treatment with said cofactor can prevent progression of kidney disease and death.
As is the case with any genetic disease, the existence of relatives with similar clinical conditions should make us think of a mitochondrial disease, although, at times, the wide fenotype variability of these disorders can make our job difficult. According to the location of the mtDNA or nDNA mutation, inheritance will be exclusively maternal and we must search for affected relatives on the mother’s side of the family, but in the case of an autosomal recessive inheritance we must look for the disease in siblings; bearing in mind that large deletions of mtDNA usually occur spontaneously. In addition, another fact that should make us suspect mitochondrial disease with renal implication, is the involvement of various organs, especially the central nervous system and skeletal muscles in a patient with proximal tubulopathy, proteinuria or distal tubulopathy. Most mitochondrial diseases, although not all, also present lactic acidosis. Therefore, the presence of elevated lactic acid in blood and urine can help. It is important to remember that not all patients with deafness and renal disease are suffering from Alport syndrome and for a differential diagnosis between this syndrome and mitochondrial diseases analysis of urinary sediment is critical.
We must not forget that mitochondrial diseases can affect any organ and the presence of symptoms in some of them may be delayed or asymptomatic, so once we have a diagnosis we must request additional studies to determine overall disease burden: heart studies (echocardiogram, Holter) eye studies (fundoscopy, visual acuity), neurological studies (elecromyogram, SNC images), endocrine studies (thyroid hormones, glycaemia, etc.), renal studies (urinary sediment, proteinuria, excretion of calcium, magnesium sodium, potassium, amino acids, glucose, etc.), ear nose and throat studies (audiometry).
There is a software, www.simulconsult.com that compares findings in a patient with more than 5000 genetic diseases, helping to make a differential diagnosis in addition to suggesting additional tests to achieve the correct diagnosis.
Conflicts of interest
The authors declare the following potential conflicts of interest.
They receive grants from: This study was partially funded by research project PI12 / 01683 of the Health Institute Carlos III-Ministry of Economy and Competitiveness.

Table 1. Most common clinical manifestations and complementary laboratory tests recommended in patients with suspected MIDD (maternally inherited diabetes and deafness)

Table 2. Review of cases of maternally inherited diabetes and deafness with renal involvement in the form of focal segmental glomerulosclerosis

Table 3. Renal diseases associated with mutations in mitochondrial deoxyribonucleic acid

Table 4. Renal diseases associated with mutations in nuclear deoxyribonucleic acid

Figure 1. Mitochondrial DNA Molecule

Figure 2. Pathological anatomy



