






The complement system is a first line of defence against infectious, tumoral or autoimmune processes, and it is constitutively regulated to avoid excessive or unspecific activation. Factor H (FH), a most relevant complement regulator, controls complement activation in plasma and on the cellular surfaces of autologous tissues. FH shares evolutionary origin and structural features with a group of plasma proteins known as FH-Related Proteins (FHRs), which could act as FH functional antagonists. Studies in patient cohorts of atypical Haemolytic-Uraemic Syndrome (aHUS), C3 Glomerulopathy (C3G), and IgA nephropathy (IgAN), have identified rare genetic variants that give rise to severe FH and FHRs dysfunctions, and are major genetic predisposing factors. These patients also have a higher frequency of a few polymorphisms whose relevance as disease risk factors is incompletely understood. In the last years, the availability of specific reagents has allowed a more precise quantitation of FH and FHRs in plasma samples from patients and controls. These studies have revealed that some aHUS, C3G or IgAN risk polymorphisms determine mild changes in FH or FHRs levels that could somehow perturb complement regulation and favour disease pathogenesis.
El sistema del Complemento protege al organismo frente a procesos infecciosos, tumorales y autoinmunes, y requiere una regulación muy estricta para evitar una activación excesiva o inespecífica. Entre los componentes reguladores del Complemento destaca el factor H (FH), que controla su activación en plasma y sobre la superficie de las células y tejidos propios. FH está relacionado evolutiva y estructuralmente con un conjunto de proteínas plasmáticas denominadas FHRs (FH-Related proteins), que podrían actuar como antagonistas funcionales de FH. Numerosos estudios realizados en pacientes de Síndrome Hemolítico-Urémico atípico (SHUa), glomerulopatía C3 (GC3), y nefropatía por IgA (NIgA) han identificado variantes genéticas raras que alteran sustancialmente la función del FH y las proteínas FHRs, y contribuyen de forma muy relevante a la predisposición genética a estas patologías. Estos pacientes presentan también una mayor frecuencia de determinados polimorfismos cuya repercusión en el mecanismo patogénico se está empezando a dilucidar. En los últimos años, la disponibilidad de reactivos específicos para cuantificar las proteínas FHRs de forma fiable en controles y pacientes, ha mostrado que algunos de los polimorfismos asociados a SHUa, GC3 o NIgA determinan cambios en los niveles plasmáticos de FH y proteínas FHRs, que podrían repercutir en la correcta regulación de la activación del Complemento y contribuir así al desarrollo de estas patologías.
The complement system is essential for the innate immune response, and it also modulates adaptive immunity because it fosters the generation of antibodies and immunological memory. In addition to immediately protecting the body against a wide variety of pathogens, complement plays a key role in eliminating immune complexes and damaged self cells, which could cause tissue damage and favour autoimmune processes.1
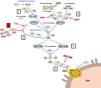
Complement is made up of more than 30 constitutively synthesised plasma or membrane proteins that exert an effector function or a regulatory function intrinsic to the system's homeostasis. The entire functioning of complement pivots around the activation process of complement component 3 (C3), one of the most abundant plasma proteins (Fig. 1). C3 activation is a proteolytic reaction by which the C3 molecule is cleaved into C3b and C3a fragments, and which can be arrived at via three pathways in which different components participate: the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP). The generation of C3b, in turn, leads to the final stage of complement activation, known as the lytic pathway.2
Complement activation and regulation.
Complement is a self-regulating system whose cornerstone is the proteolytic activation of the C3 component. 1) Spontaneous hydrolysis of some C3 molecules leads to basal activation of the alternative pathway (AP). 2) Complement can be activated more intensely through the classical pathway (CP) and the lectin pathway (LP), when the C1q or MBL/Ficolin components recognise soluble antigen-antibody complexes, or carbohydrate molecules present on certain surfaces (damaged self cells or microorganisms). This activation leads to the formation of a C3 convertase (C4bC2b), which cleaves C3 into C3b and C3a fragments. 3) The generation of C3b gives rise to another C3 convertase (C3bBb) and to the amplification of the basal activation of the AP, which then becomes the most effective mechanism of complement activation. 4) The next step is the activation of the C5 component, generating C5b and C5a fragments. 5) The last stage is the so-called Lytic Pathway, which gives rise to the formation of a multimolecular complex (MAC, Membrane Attack Complex) that inserts into the membrane of the activating surface and forms pores that lyse it due to osmotic imbalance. 6) The complement activation process is regulated in all its stages by multiple proteins (represented in red) that prevent it from being activated on self cells and tissues; AP regulation largely depends on the regulator Factor H and its FHL-1 isoform. The precise role of FHR proteins (structural homologues of FH) in the complement activation-regulation process is not fully understood. For complement nomenclature, see the references.49,50
The biochemical characteristics of the complement activation process make it an extremely fast and effective tool for defence against pathogens, but it can also be harmful to the host. Excessive activation can lead to a rapid consumption of components that facilitates a new invasion by pathogens, and causes an exacerbated inflammatory response. Moreover, complement activation products can be deposited on self cells and damage them. In physiological situations, these adverse consequences are avoided thanks to the fact that each and every one of the different stages of the complement activation process are controlled by regulatory components, some present in plasma and others located on the surface of most self cells and tissues. Consequently, for complement to be effective and specific, and not to cause autologous damage, balance between its activation and regulation is essential.3
Regulation of the complement AP is especially relevant. This activation pathway, evolutionarily the oldest, is permanently activated in plasma due to spontaneous hydrolysis of an internal thioester bond in some C3 molecules. Normally, this basal activation of the AP does not exceed a detrimental threshold, thanks to complement components that regulate it. For this reason, any situation, whether genetic or acquired, that alters the regulation of the AP can cause autologous damage by the complement, with the kidney being one of the most vulnerable organs.4 Kidney diseases commonly associated with dysregulation of the complement alternative pathway are atypical haemolytic uraemic syndrome (aHUS), complement 3 glomerulopathy (C3G) and IgA nephropathy (IgAN). Many of these patients have pathogenic genetic variants or circulating autoantibodies that affect the normal functioning of Factor H (FH), the key regulator of the complement AP, and which is capable of controlling C3 activation both in plasma and on self cell surfaces; and no less relevant is the existence of common genetic variants (i.e., polymorphisms) in the FH gene that also contribute to the development and/or clinical expression of these diseases.5 The first part of this review will summarise the pathogenic variants and polymorphisms that affect the FH-dependent regulation of the complement AP, before subsequently looking at the repercussions that FH levels and their homologous proteins, factor H-related proteins (FHRs), can have on kidney disease.
Family of FH and FHR proteinsFH is a 150-kDa plasma glycoprotein, composed of 20 structural domains called SCRs (Short Consensus Repeats) or CCPs (Complement Control Proteins). Two functionally very relevant regions can be distinguished in the FH molecule: the N-terminal region (SCRs 1–4), which controls its ability to regulate complement activation in plasma, and the C-terminal region (SCRs 19–20), which allows FH to transiently bind to self cell surfaces and protect them from indiscriminate damage by complement.6
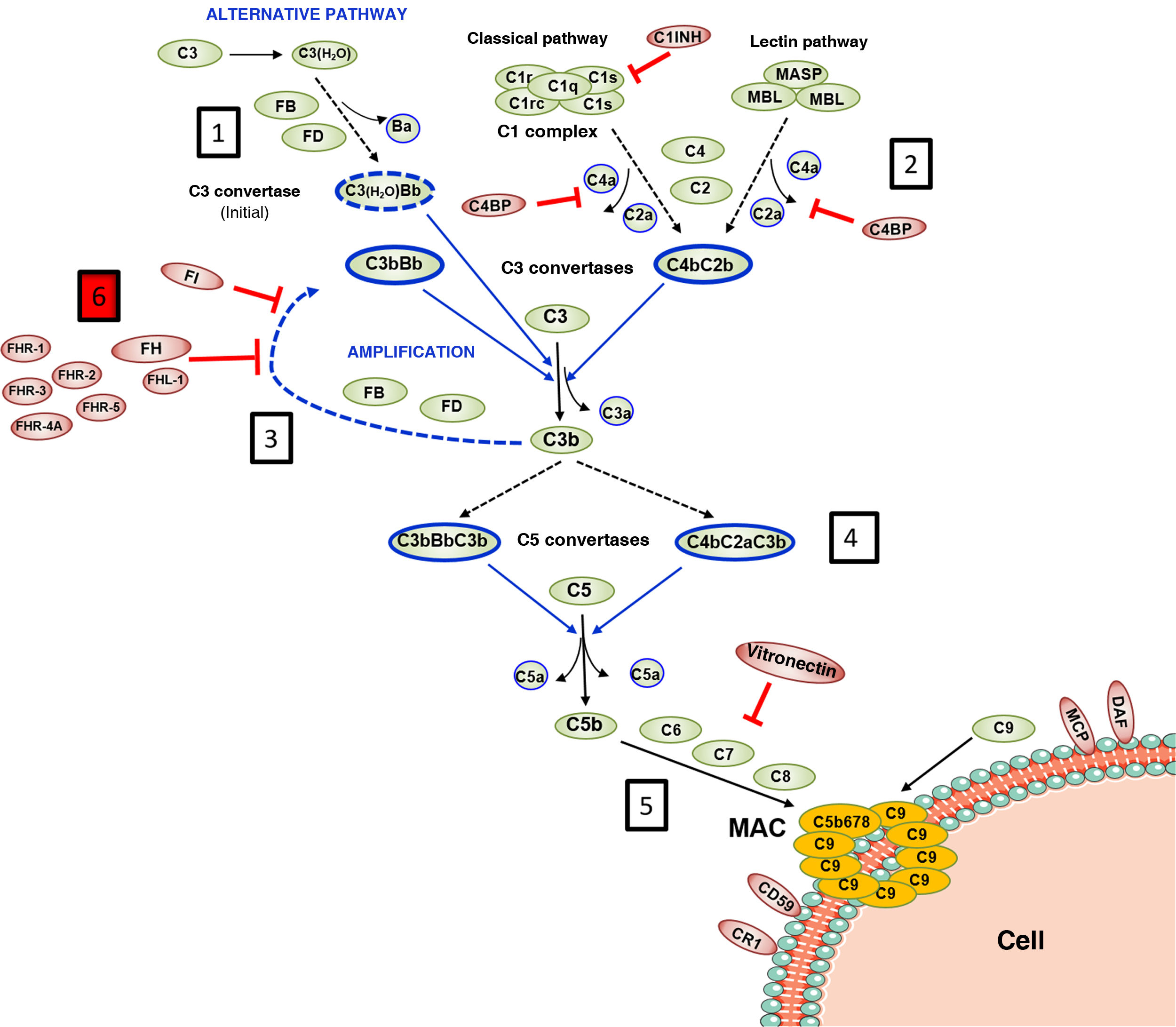
FH is encoded by the CFHgene, which also generates a variant of alternative splicing named Factor H-like protein 1 (FHL-1). The FHL-1 protein is composed of only the first seven SCR domains of FH, followed by a tail of four amino acids; as a result, FHL-1 can regulate complement like FH, but cannot bind to self cell surfaces.7 Along with the CFH gene, the five CFHRs genes are found in the Regulators of Complement Activation (RCA) gene cluster of chromosome 1, which encode the five FH-homologous proteins collectively called FHRs.8 The CFH-CFHRs genes have very similar exonic and intronic regions, reflecting a shared evolutionary history, and characterised by a succession of gene duplication events (Fig. 2a). This common origin explains the high structural similarity of FH and FHR proteins, which are also composed entirely of SCR domains, some of which reach 100% amino acid identity with the homologous domains in FH. The distinct molecular size of FH and the FHR proteins allows all of them to be easily identified by Western-blot analysis in a sample of plasma or serum from the patient (Fig. 2b).
Family of factor H and FHR proteins.
A) RCA (Regulator of Complement Activation, 1q32−33) gene cluster region that contains the CFH gene and the five CFHRs genes. All genes have the same 5'-3' orientation. Letters A, B, C and D denote exon-intron groups homologous to each other. B) Identification of FH, FHL-1 and the five FHR proteins from normal human serum by Western-blot analysis with two antibody preparations with different specificity. All proteins are made up of 4–20 homologous globular domains; domains with the same colour are the most similar to each other and are encoded by A, B, C or D duplications of the respective genes.
FHR proteins can interact with C3b and bind to FH ligands located on the cell surface. However, no FHR protein has SCR domains homologous to the N-terminal region of FH, so they cannot regulate plasma complement activation in the same way, nor protect self cell surfaces against autologous complement. These characteristics led to the proposal that FHR proteins acted as FH deregulators,9 but their precise role is not well characterised and is disputed.10 In this sense, it cannot be ruled out that some FHR proteins have certain complement regulatory activity,11,12 but it has also been demonstrated that they can act as activators. This is the case for the proteins FHR-1, FHR-4 and FHR-5, which can bind to C3b, promote the formation of C3 convertase and activate the AP, and can also activate the CP by binding to the pentraxins PTX3 and C-reactive protein.13–17
Since FHR proteins deregulate FH and/or directly potentiate complement AP activation, their physiological function is antagonistic to that of FH. Therefore, genetic variants that alter the function of FH and FHR proteins, or that alter their plasma levels, can deregulate the AP and cause predisposition to the development of kidney disease.
Genetic variants CFH/CFHRs in aHUS, C3G and IgANAs mentioned above, the functional balance between FH and FHR proteins contributes to maintaining complement AP homeostasis and preventing the development of kidney diseases such as aHUS, C3G or IgAN. Studies conducted in different cohorts of aHUS and C3G patients have identified pathogenic variants of FH and FHR proteins that disrupt AP regulation, and these are detailed in recent reviews.18,19 Some pathogenic variants have also been described in IgAN patients.20,21
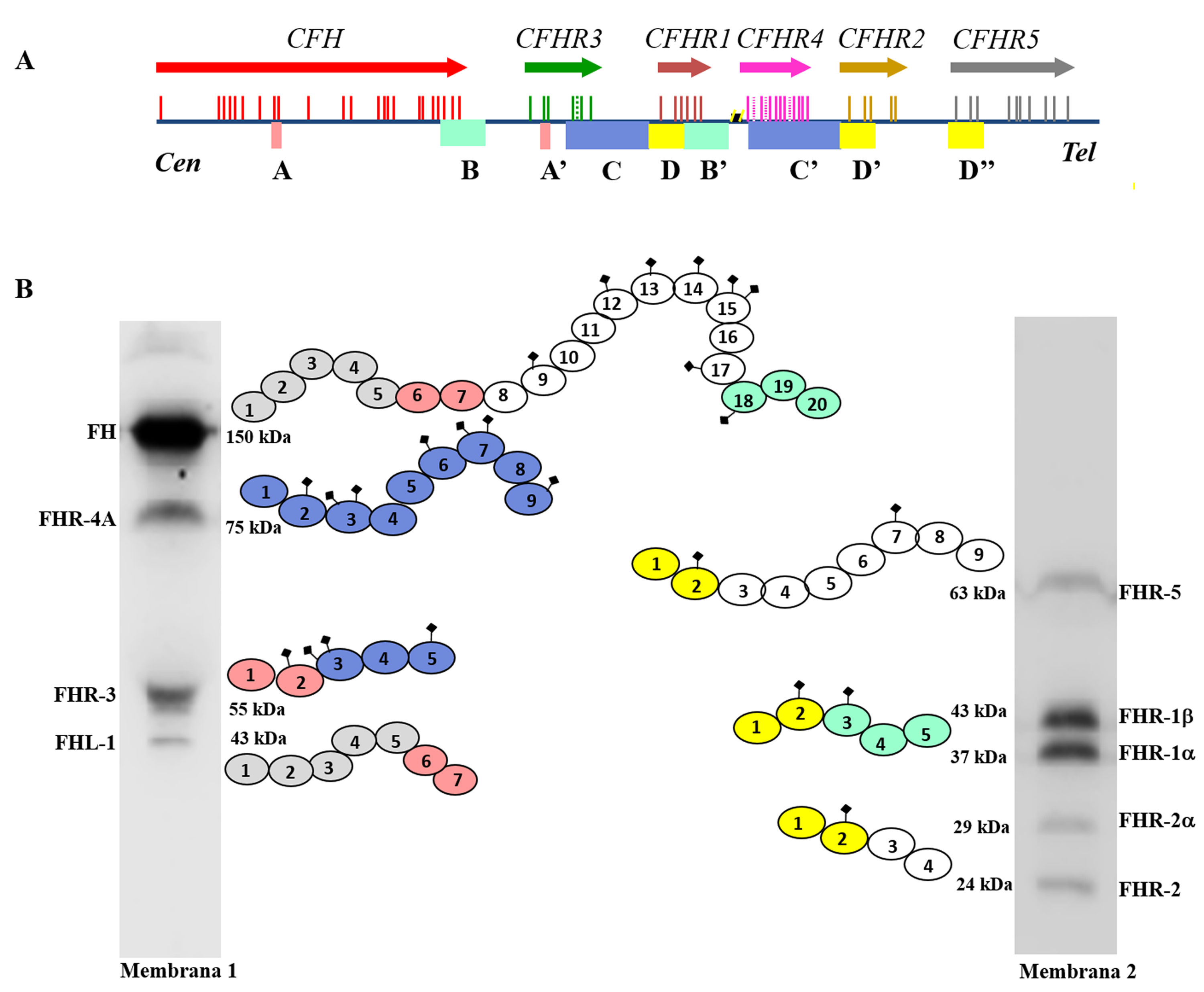
Many of these pathogenic variants result from the existence of exon-intron duplications in the CFH-CFHRsregion, which make it very prone to gene conversion events and unequal homologous recombination that cause mutations, duplications, deletions or hybrid genes. Most of these abnormal rearrangements cannot be detected by standard DNA sequencing, so techniques capable of determining the number of copies of the region suspected of being duplicated or absent are needed, or even much more specific techniques that are only performed in some research laboratories and are time consuming. Some of these abnormal rearrangements cause complete deficiency of one or more proteins, while others generate proteins that have a different molecular size from the native proteins. These two eventualities are quickly detected by a simple Western-blot analysis (Fig. 3), which, in addition, provides valuable information for designing specific genetic strategies to characterise the underlying genetic defect.
Screening of deficiencies and abnormal forms of FH and FHR proteins in aHUS and C3G patients by Western-blot.
A) FH and FHRs patterns in a healthy individual (SHN) and in five aHUS and C3G patients who have complete deficiency of one or two proteins; dashed squares denote the position of the missing protein. The differences in the intensity of other bands reflect the fact that there is significant interindividual variability in FH and FHR protein levels, the pathophysiological relevance of which is not sufficiently clear.
B) FH and FHRs patterns in two patients showing abnormal bands (marked with an asterisk), whose size does not correspond to that of any native protein. Additional WB analysis and genetic studies identified that the abnormal band from patient 6 corresponds to a short isoform of FHR-3, and the abnormal band from patient 7 corresponds to a partially duplicated form of FHR-1.
In addition to rare pathogenic variants, the CFH-CFHRs region contains a multitude of polymorphisms (i.e. genetic variants with a frequency in the population greater than 1%), some of which are associated with kidney disease (Table 1). Of these polymorphisms, the deletion of the CFHR3 and CFHR1 (ΔCFHR3-CFHR1) genes is particularly interesting, which, when homozygous, is associated with a risk of aHUS22 and protection against IgAN.23 In total, 80% of aHUS patients who develop anti-FH autoantibodies are homozygous for the variant ΔCFHR3-CFHR1 and therefore lack the FHR-3 and FHR-1 proteins.
Common genetic variants in the CFH-CFHRsregion and kidney disease.
| Variant | Functional consequence | Association | Reference |
|---|---|---|---|
| CFH(H1) | Contains the His402 variant that decreases FH binding to different membrane carbohydrates | C3G risk | 24 |
| CFH(H2) | Contains the Ile62 variant that increases FH regulatory activity. | C3G and aHUS protection | 24 |
| CFH(H3) | Associated with lower levels of FH | aHUS risk | 25 |
| CFH(H4) | Associated with the ΔCFHR3-CFHR1variant | aHUS risk (HOM) | 46 |
| Higher FH levels | |||
| ΔCFHR3-CFHR1 | Associated with anti-FH autoantibodies | aHUS risk (HOM) | 26 |
| FHR-1 deficiency favours FH regulation | IgAN protection | 23 | |
| CFHR3*B | Associated with higher levels of FHR-3 | aHUS risk | 27 |
| CFHR1*B | Contains the Tyr157-Val159-Gln175 sequence, which could increase the competition of FHR-1 with FH. | aHUS risk | 26 |
| CFHR4*B | Contains the Pro405 variant, but its functional impact is unknown. | aHUS risk | 28 |
| CFHR5Ser62 | Affects the interaction of FHR-5 with C3b and could influence complement activation | C3G risk | 29 |
The CFH gene contains single nucleotide polymorphisms, (SNPs) that are found in linkage disequilibrium and define different haplotypes, the most common being CFH(H1), CFH(H2), CFH(H3) and CFH(H4). The CFH(H1) haplotype, which contains the FH variantHis402, is associated with a risk of C3G, while the CFH(H2) haplotype, which includes the FH variantVal62, is considered a protective factor against aHUS and C3G.24 The CFH(H3) haplotype is associated with a risk of aHUS and with lower FH levels.25 Finally, haplotype CFH(H4) is in linkage disequilibrium with the deletion ΔCFHR3-CFHR1, and it is more common in aHUS patients with anti-FH autoantibodies.
The CFHR1 and CFHR3 genes also have several SNPs in linkage disequilibrium that configure two major alleles: CFHR1*A/CFHR1*B26 and CFHR3*A/CFHR3*B.25 The CFHR1*B and CFHR3*B alleles are associated with a risk of aHUS. The CFHR3*B allele determines higher levels of the FHR-3 protein,27 while the CFHR1*B allele does not seem to be related to FHR-1 protein levels; in this case, its association with aHUS is probably due to the fact that it has greater amino acid identity with FH and therefore could compete more effectively for binding to the same ligands.
Interestingly, the aHUS risk haplotype CFH(H3) occurs mostly on the same chromosome as the CFHR3*B and CFHR1*B alleles, giving rise to extended haplotype CFH(H3)-CFHR3*B-CFHR1*B, which probably determines an increase in the relative levels of FHR-3/FH that could favour the development of aHUS. Haplotype CFH(H1), for its part, usually occurs with the CFHR3*A and CFHR1*A alleles, forming the extended haplotype CFH(H1)-CFHR3*A-CFHR1*A, which is predominant in the healthy control population.
In the CFHR4 gene, a polymorphism apparently associated with aHUS and that does not seem to be related to FHR-4A protein levels has also been described,28 and the CFHR5 gene contains several polymorphisms that are associated with increased risk of C3G.29
In IgAN patients, the CFH-CFHRs region has not been characterised in the same detail as in aHUS and C3G patients. For this reason, the possibility that there may be other polymorphisms associated with IgAN, apart from the aforementioned ΔCFHR3-CFHR1 deletion, cannot be ruled out.
FH levels and FHR proteins in controls and patientsThe activity of the complement AP can also be affected by changes in the physiological levels of FH and FHRs, as shown by the studies that we will comment on in this section, in which we will also analyse the polymorphisms in the CFH-CFHRs region that affect protein levels. It is important to note that most of these studies express protein levels in μg/mL; since the molecular size of FH (150kDa) is greater than that of any of the FHRs (between 24kDa and 75kDa), in order to correctly interpret the changes in FH and FHRs plasma levels, the concentrations of all of them must be compared on a molar basis (Fig. 4).
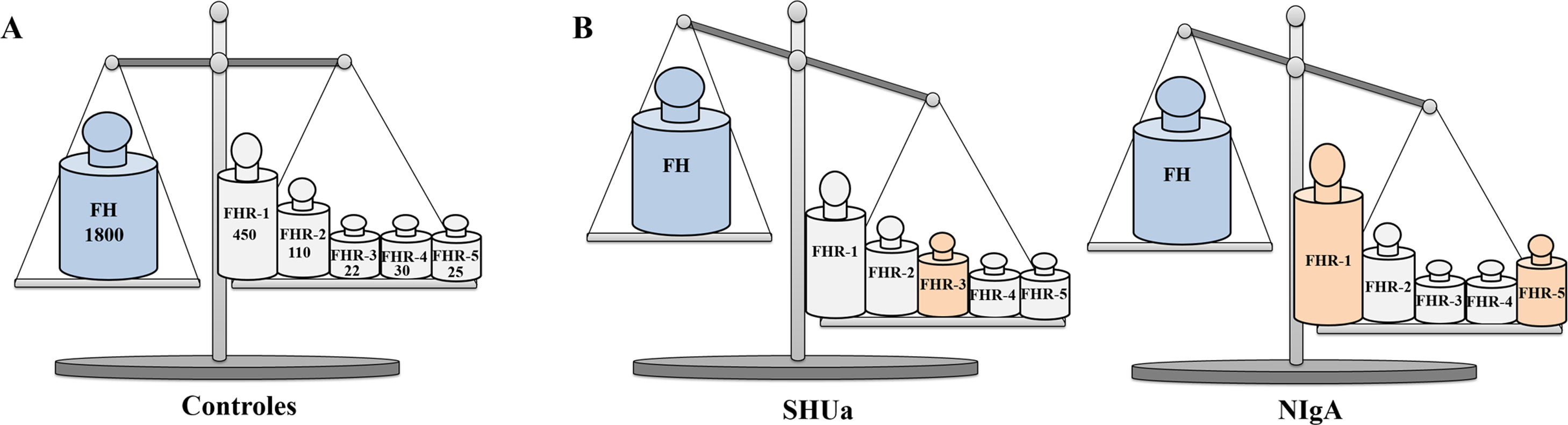
Pathophysiological relevance of FH and FHR protein plasma levels.
Graphical representation of FH and FHR protein levels in controls, aHUS patients and IgAN patients. A) In healthy controls, FH and FHR protein levels are in functional equilibrium. The numbers underneath the name of each protein express its mean concentration on a nanomolar basis,33,40,43 so they are equivalent to the number of molecules of each of them in a given volume of plasma. B) Small increases in the concentration of any FHR protein can cause an imbalance in the regulation of the complement AP by FH, and favour the development of kidney diseases; FHR-3 levels increase in some aHUS patients,27 while in some IgAN patients, FHR-1 and FHR-5 levels increase.21,34
FH is a relatively abundant protein in plasma, with a wide concentration range (116−562μg/mL), which is 62% genetically determined, increases slightly with age, and decreases with tobacco use.30 Mean values in adult controls are approximately 250μg/mL,21,31–35 similar to those observed in paediatric controls36 and greater than in neonates.37 Levels of FHL-1 (the short isoform of FH), on the other hand, are between 3 and 40 times lower than those of FH, on a molar basis.7,38
The high similarity between FH and FHRs has been a handicap when it comes to generating specific antibodies to quantify each of these proteins by ELISA assays. For this reason, other techniques have been used (semiquantitative Western-blot, mass spectrometry) that have provided very disparate results.10 In recent years, several research groups have generated specific antibodies for all the FHR proteins, facilitating the more reliable determination of their plasma concentration. Commercial antibodies currently exist for all the FHRs, except the FHR-3 protein, meaning that it is possible to design specific ELISA assays to quantify them. However, the fact that there is no common protocol, coupled with the lack of reference reagents, means that the levels obtained in these tests are sometimes very different. An added complication is the fact that FHR-1, FHR-2 and FHR-5 are capable of forming dimeric complexes. This peculiarity is due to the fact that the SCR1-SCR2 domains of these three proteins are very similar to each other and contain amino acids that allow them to form homodimers and heterodimers that have a greater capacity to compete with FH.39 The quantification of all these forms has been addressed in a study carried out in Dutch controls, according to which there would be no monomeric forms of these proteins circulating in plasma.40
FHR-1 and FHR-3 protein levels are determined, in the first instance, by the existence of the ΔCFHR3-CFHR1 genetic variant, since individuals homozygous for this variant do not have any copies of the CFHR1 and CFHR3genes and heterozygous individuals only have one. As the frequency of the ΔCFHR3-CFHR1 genetic variant varies considerably between populations, it is a very relevant genetic trait when comparing FHR-1 and FHR-3 levels between different cohorts of healthy controls, or between controls and patients of different ethnicities. As such, 5% of European Caucasian individuals do not have any copies of the CFHR1 and CFHR3genes, and this frequency rises to 33% in individuals from Nigeria, while in the Japanese and South Americans it is practically zero.41 In this section we will refer to the data obtained in Caucasian populations.
FHR-1 levels in individuals with a single copy of the CFHR1 gene (i.e. heterozygous for the ΔCFHR3-CFHR1 variant) are approximately half the level in those with two copies of the gene (61μg/mL vs 122μg/mL).21 These results are reasonably consistent with the levels of 95μg/mL observed in another European control population, in which the number of copies of the ΔCFHR3-CFHR1 variant was not taken into account.34 However, the study in Dutch controls, which also discriminates between FHR-1/1 homodimers and FHR-1/2 heterodimers, found total FHR-1 values about 10 times lower.40 FHR-3 protein plasma levels are well below those of FH and FHR-1 but, as might be expected, they are also about twice as high in adult controls who have two copies of the CFHR3 gene than in those who only have one (0.83μg/mL vs 0.38μg/mL).33 In FHR-3 quantification, however, an additional genetic component must be taken into account, since the CFHR3*B allele gives rise to higher protein levels than the CFHR3*A allele.27 Therefore, the levels of FHR-3 in plasma will ultimately depend on the number of copies of the CFHR3*A and CFHR3*B alleles that each individual has, which explains the great variability of FHR-3 levels (between 0.14 and 1.16μg/mL) observed in a small Spanish control cohort in which ΔCFHR3-CFHR1, CFHR3*A and CFHR3*B variants were characterised.27 In paediatric controls, FHR-1 levels are slightly lower than in adults, while no differences in FHR-3 levels are observed.36
The mean FHR-2 protein plasma concentration of adult controls is about 6μg/mL, which corresponds mainly to FHR-1/2 heterodimers (5.2μg/mL) and to a much lesser extent (0.8μg/mL) to FHR-2/2 homodimers.40 FHR-2 levels in paediatric controls are very similar to those seen in adults.36 No polymorphisms of the CFHR2 gene have been described that affect the levels of protein expression.
The FHR-4A protein is the major isoform of the CFHR4 gene, which also gives rise to a product of alternative splicing called FHR-4B, smaller than the FHR-4A isoform.42 The first quantifications did not distinguish the FHR-4A and FHR-4B isoforms, and estimated mean protein levels of 25.4μg/mL.14 The development of specific antibodies has made it possible to quantify the two isoforms individually.43 The results demonstrate that levels of the FHR-4A isoform are 2.55μg/mL (10 times lower than previously estimated), while the FHR-4B isoform is not detected in plasma, raising reasonable doubts as to whether it is actually secreted. FHR-4A levels do not appear to be associated with age, but are slightly lower in paediatric controls than in adults.36 A polymorphism in intron 1 of the CFHR4 gene has recently been described that is associated with higher levels of the FHR-4A isoform.35
The FHR-5 protein, like FHR-1 and FHR-2, also has two dimerisation domains, but seemingly only circulates in plasma as FHR-5/5 homodimers. Mean FHR-5 levels in Dutch adult controls are 1.66μg/mL,40 somewhat lower than those previously reported in other cohorts, which range between 2.5 and 5.5μg/mL.34,44,45 FHR-5 levels are lower in children under three years of age; above the age of three, they reach practically the same values as in adults.36
How do FH and FHR proteins levels vary in aHUS, IgAN and C3G patients? As mentioned above, some polymorphisms that directly affect protein levels have different frequencies in patients and controls. This is the case with haplotype CFH(H3), which is associated with lower levels of FH and higher levels of FHR-3,25 with haplotype CFH(H4), which is associated with higher levels of this protein,46 and with the ΔCFHR3-CFHR1deletion, which causes deficiency of FHR-1 and FHR-3.
On the other hand, several studies in aHUS and IgAN patients suggest that FH and FHRs levels also change during the course of the disease. As such, FHR-1 and FHR-5 levels, and the FHR-1/FH and FHR-5/FH ratios, are higher in IgAN patients than in the corresponding control populations, and there is a direct correlation between FHR-5 levels and disease progression.21,34,45 In the same sense, some IgAN patients have a greater FHR-5 deposition in the glomeruli, which correlates with histological damage and serves as a prognostic marker.47 Changes in FHR-5 levels have also been observed in some patients with aHUS associated with Streptococcus pneumoniae infection, who show higher levels of this protein in the plasma samples obtained during the first few days of the infectious process than in the samples obtained from patients in remission.48 This probable influence of clinical course on FH and FHRs levels would also explain why the FHR-3 levels in patients from the Spanish aHUS cohort are higher than in healthy individuals who have the same CFHR3 genotype.27 To address these questions, it will be necessary to compare FH and FHR protein levels in serial samples of patients, obtained at onset and on remission. There are no publications documenting changes in FH and FHRs levels in C3G patients. However, preliminary results from our group show that most patients have elevated levels of some FHR proteins, and this probably also applies to other C3G cohorts.
Another issue that seems relevant to us is that in most of the studies carried out in controls and patients, the absolute levels of FH and FHRs have been compared, but only in some have their relative levels, such as the FHR-1/FH or FHR-5/FH ratios, been calculated. Increased levels of a certain FHR protein may be relevant in an individual with constitutively low levels of FH, while in an individual with a high concentration of FH, this same increase may not be significant. In our opinion, changes in the relative concentrations of FH and FHR proteins can be as or more informative than changes in their absolute levels.
In summary, although the quantification of the FHR proteins could not be carried out reliably until a few years ago, it is known that the levels of some of them, in particular FHR-1 and FHR-3, are determined to a large extent by polymorphisms in the CFH-CFHRs region. On the other hand, data obtained in aHUS and IgAN patients reveal that the levels of some FHR proteins and the FH/FHRs ratio change over the clinical course of the disease. These data suggest a relevant contribution of FH and FHRs levels in the pathogenesis of diseases related to the deregulation of the complement AP, which will be further profiled as our understanding of the functional activity of these proteins and the mechanisms that determine their expression levels improves.
Key points- •
Factor H (FH) regulates the alternative complement pathway and prevents it from being nonspecifically activated on self cells and tissues.
- •
FHR proteins are structurally very similar to FH, but it is likely that they behave as functional antagonists of FH and enhance complement activation.
- •
FH and FHR protein levels are partially determined by polymorphisms in the CFH-CFHRsregion, whose frequency varies between different populations.
- •
Small changes in FH and FHR protein levels can unbalance complement regulation at the local or systemic level and favour the development of kidney diseases such as aHUS, C3G or IgAN.
IGD has a contract funded by the Autonomous Community of Madrid (Complement II-CM; S2017/BMD-3673) and by the Fundación Senefro [Senefro Foundation] (http://www.senefro.org/; Research Grants 2018). This article was funded by the Instituto de Salud Carlos III (ISCIII) [Charles III Institute of Health] and the European Regional Development Fund (project PI19/00970 a PS-C).
Conflicts of interestThe authors declare that they have no conflicts of interest.
We would like to thank Dr Margarita López Trascasa and Dr Fernando Corvillo, members of the IdiPAZ Complement Research Group, for reading the manuscript and their comments.
Please cite this article as: Gómez Delgado I, Sánchez-Corral P. Contribución de variantes funcionales y cuantitativas del Factor H y las proteínas FHRs (Factor H-Related proteins) del Complemento en patología renal. Nefrologia. 2022;42:280–289.



