
Gout is recurrent inflammatory arthritis caused by the deposition of monosodium urate crystals in the joints. The risk factors that predispose to suffering from gout include non-modifiable factors such as gender, age, ethnicity and genetics, and modifiable factors such as diet and lifestyle. It has been shown that the heritability of uric acid levels in the blood is greater than 30%, which indicates that genetics play a key role in these levels.
Hyperuricaemia is often a consequence of reduced renal urate excretion since more than 70% is excreted by the kidneys, mainly through the proximal tubule.
The mechanisms that explain that hyperuricaemia associated with reduced renal urate excretion is, to a large extent, a proximal renal tubular disorder, have begun to be understood following the identification of two genes that encode the URAT1 and GLUT9 transporters. When they are carriers of loss-of-function mutations, they explain the two known variants of renal tubular hypouricaemia.
Some polymorphisms in these genes may have an opposite gain-of-function effect, with a consequent increase in urate reabsorption. Conversely, loss-of-function polymorphisms in other genes that encode transporters involved in urate excretion (ABCG2, ABCC4) can lead to hyperuricaemia.
Genome-wide association study (GWAS) methods have made it possible to locate new gout-related loci associated with reduced renal urate excretion (NIPAL1, FAM35A).
La gota es una artritis inflamatoria recurrente provocada por el depósito de cristales de urato monosódico en las articulaciones. Entre los factores de riesgo que predisponen a padecer gota se encuentran aquellos no modificables como sexo, edad, raza y genética y los modificables como dieta y estilo de vida. Se ha indicado que la heredabilidad de los niveles de ácido úrico en sangre es superior al 30%, lo que indica que la genética tiene un papel clave en dichos niveles.
La hiperuricemia es a menudo una consecuencia de la reducción de la excreción renal de urato, ya que más del 70% se excreta por el riñón, principalmente, por el túbulo proximal.
Los mecanismos que explican que la hiperuricemia asociada a la reducción de la excreción renal de urato es, en gran medida, una tubulopatía proximal, se han empezado a conocer al saberse la existencia de dos genes que codifican los transportadores URAT1 y GLUT9 que, cuando son portadores de mutaciones de pérdida de función, explican las dos variantes conocidas de hipouricemia tubular renal.
Algunos polimorfismos presentes en esos genes pueden tener un efecto contrario de ganancia de función, con la consecuencia de un incremento en la reabsorción de urato. A la inversa, polimorfismos de pérdida de función en otros genes que codifican trasportadores implicados en la excreción de urato (ABCG2, ABCC4) favorecen la hiperuricemia.
Los métodos de asociación genómica amplia (GWAS) han permitido localizar nuevos locus relacionados con gota asociada a reducción de la excreción renal de urato (NIPAL1, FAM35A).
“Whoever suffers from gout or bad joints deserves it; because they eat what suits their appetite and they don't have a rule to use good food : and more than that they don't exercise (sic)” (Luis Lobera)1
Gout is a common disease that causes acute episodes of arthritis associated with persistent hyperuricemia and monosodium urate crystal deposition in joints, soft tissues, and kidneys. It is a condition known since ancient times. At the time of Hippocrates (5th century BC) it was known by the name of podagra (from pod [o]: foot and ágrā: hunting, traps, that is, “traps that catches the foot”). Instead, the word gout derives from the Latin gutta (gutta quam podagram vel artiticam vocant - gout is called podagra or arthritis). It was used by doctors in the Middle Ages to designate the disease "caused by a humor that apparently flowed drop by drop, especially in the joints of the foot." Many historical figures are known to have suffered from the disease, such as King Carlos I of Spain (the demonstration of the existence of uric acid crystals [UA] in one of his little fingers was carried out by Julián de Zulueta et al. in 2006).2 Luis Lobera included gout among the four illnesses of the court together with the catarrh or « rheuma », bladder calculosis and «French disease» or «bubas» (syphilis).1 Thomas Sydenham (1624-1689), the famous English doctor affected by the disease, made a proverbial clinical description of acute crises.3
Anton van Leeuwenhoek, the well-known innovative and notable merchant microscopist (1632-1723) was, apparently, the first to describe the appearance of the crystals that make up a gouty tophus, although their chemical composition was unknown at the time: «I observed the solid material that to our eyes resembles chalk, and I saw with great astonishment that my first opinion was mistaken, since it consisted of nothing more than small long transparent particles, many pointed at both ends» (1679).4
The scientific history of gout began in 1776 when the Swedish chemist Carl Wilhelm Scheele (1742-1786) discovered UA as a component of a kidney stone5 William Hyde Wollaston (1766-1828), a British physicist and chemist, described in 1797, at the Royal Society in London, the material obtained from a tophi from his own ear and stated that it was made up of lithic acid (from lithos, stone) and an alkaline mineral.6 The French chemist Antoine de Fourcroy (1755-1809) named it uric acid.
In the mid-nineteenth century, Alfred Baring Garrod (1819-1907) carried out experiments in which he demonstrated the existence of a greater amount of UA in the blood of gouty patients and wrote that «the deposited urate of soda may be looked upon as the cause, and not the effect, of the gouty inflammation».7
One of the secondary causes of gout has been known for a long time. We refer to the saturnine gout that was delimited by the English doctor William Musgrave (1657-1721).7,8 Lead poisoning must have been very common in the past.9 This element was present in many objects used by humans such as water pipes, wine containers, kitchen pots and paintings. It has been reported that characters such as Ludwig van Beethoven and Benjamin Franklin may have suffered lead poisoning, as well as painters such as Caravaggio, Francisco de Goya and others.10
Etiology of hyperuricemiaHyperuricemia can be classified into two basic etiological subtypes, one due to renal overload (renal overload) and the other due to reduced renal excretion of UA (renal underexcretion).
Hyperuricemia secondary to UA overproduction may be a consequence of increased purine catabolism or ATP degradation. Among the genetic causes of excess UA production should be mentioned the Lesch-Nyhan and Kelly- Seegmiller syndromes, hyperactivity of phosphoribosylpyrophosphate-synthetase (PRPP - synthetase) and some glycogenosis. Increased purine catabolism occurs in hematological and neoplastic diseases and in extensive psoriasis, as well as in the presence of destruction of a large number of cells during chemotherapy treatment. The competition of UA with other organic acids for renal excretion explains the hyperuricemia associated with diabetic ketoacidosis, alcoholic overdose11,12 or fasting states. The dietary causes of increased UA are, excess intake of drinks sweetened with fructose/sugar,13 dairy products14 and foods rich in purines,13,15 as well as lack of physical exercise.16 Among drugs causing elevation of UA, the use of diuretics is a frequent cause of hyperuricemia17; through volume depletion they lead to decreased glomerular filtration rate, increased tubular reabsorption, and possibly reduced tubular secretion. Other drugs such as low-dose aspirin, nicotinic acid, ethambutol, cyclosporine, and pyrazinamide18 interfere with renal excretion of UA.
It is noteworthy that in recent years a situation has been described that resembles hyperproduction of extrarenal origin without being so; we refer to some cases in which there is a decrease in the intestinal excretion of UA19 [due to this finding, the previous classification type of “overproduction” of UA was changed to the current concept of hyperuricemia due to “renal overload”]. Also, it is necessary to remember that there are causes of hyperuricemia in which the two mechanisms are combined, such as what happens in the case of some neoplasms in which there is an overproduction of purines due to the effect of tumor lysis together with a decrease of the excretion of AU related to the nephrotoxicity of chemotherapeutic agents.20
But, in this review, what interests us are the causes of the reduced renal excretion of UA. For this, it is necessary to review the transport systems that intervene in the tubular handling of UA.
At this time, it should be remembered that at physiological pH (7.40), about 98% of the UA is in the form of urate (anionic form) and when the urinary pH is less than 5.5, it is in the non-dissociated form.
Renal tubular handling of uric acid. First findings. The Four Component HypothesisIn 1950, Berliner et al. tried to find an explanation for the fact that the clearance of UA could be greater than that of creatinine. To elucidate this “incomprehensible phenomenon”, these authors induced hyperuricemia by means of lithium carbonate overload in a group of healthy subjects. Studying inulin and urate clearance, they concluded that urate is excreted by glomerular filtration and that there was active tubular reabsorption.21 That same year, Praetorius and Kirk described a patient with marked hypouricemia in whom UA clearance was higher than creatinine clearance. This led them to assume that the individual's kidney was secreting AU. It was the first case of tubulopathy described later as renal tubular hypouricemia.22
The existence of a tubular secretion of UA was proposed in 1957 when Gutman and Yu studied 300 patients with gout and concluded that a reduction in tubular secretion of UA would explain the reduction in uricosuria.23 Four years later, these same authors published their three-component theory, namely, urate is filtered in the glomerulus, it would be actively reabsorbed in the proximal tubule and then secreted into the tubular lumen.24 In the early 1970s, Diamond and Paolino, through the sequential use of various uricosuric agents in healthy subjects, pointed out the existence of urate reabsorption that would be located in a place distal to the secretion (postsecretory reabsorption).25 The hypothesis of the three components had been transformed into that of the four components in such a way that the filtered urate would be reabsorbed almost entirely in the proximal tubule, with 0-2% of the filtrate UA remaining in the tubular lumen. Subsequently, a phase of tubular secretion would occur that increases the amount of urate in the tubule up to a 50% of the amount initially filtered. Finally, a tubular reabsorption would occur again in an amount quantified in 80% of the secreted. This would explain why the amount of UA excreted in the urine is approximately 10% of the amount of filtered urate. In this way, the defects in the tubular management of the UA associated with hypouricemia could be caused by defects in both presecretory and postsecretory reabsorption, isolated or combined, or by an increase in its tubular secretion. Knowledge about the metabolism of UA did not change until the arrival of new advances resulting from the application of molecular biology techniques. As a consequence of these advances, several transporters were identified that have demonstrated the complexity of urate ion handling by the proximal tubule. However, at present, it has not yet been possible to fit and link the activity of these transporters with the hypothesis of the four components.
Proximal tubular transporters involved in urate reabsorptionThe urate-anion exchanger URAT1 (urate-anion transporter 1) that reabsorbs filtered urate, was identified by Enomoto et al. in 2002.26 It is located on the apical membrane of proximal tubular cells and is encoded by the SLC22A12 gene located on chromosome 11q13. URAT1 belongs to the family of organic anion transporters (organic anion transporter [OAT]). In the human kidney, urate is transported by means of URAT1 through the apical membrane of the proximal tubular cells, which is exchanged with anions that are transported towards the tubular lumen to maintain an adequate electrical balance (Fig. 1).
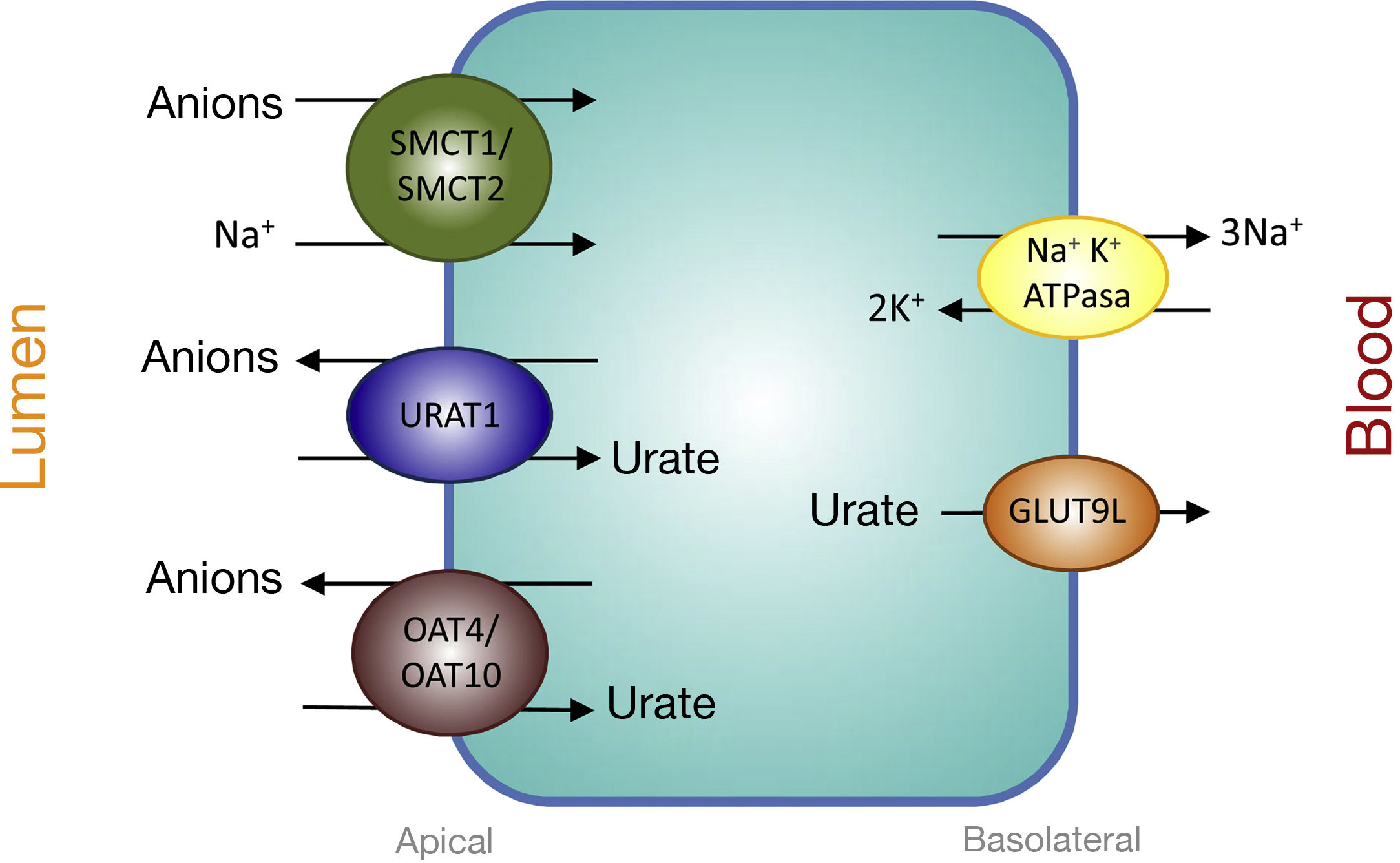
Urate reabsorption in epithelial cells of the proximal tubule. Urate reabsorption at the apical membrane of proximal tubule cells is mediated by transporters URAT1, OAT4, and OAT10. Its action is favored by an increase in the intracellular concentration of organic anions due to the action of the transporters SMCT1 and SMCT2. At the basolateral membrane, GLUT9L transports urate out of the cell into the blood.
Evidence that URAT1 plays an important role in renal urate management comes from the identification of inactivating URAT1 mutations in patients with renal tubular hypouricemia type 1.26–28 These patients are characterized by low levels of UA (between 0.5 and 1.5mg/dl), a high fractional excretion (between 20 and 90ml/100ml GFR) and an attenuated response of uricosuria to probenecid and pyrazinamide.27 Also, both losartan and benzbromarone exert their uricosuric action by inhibiting the action of URAT1.
The truth is that the tubular reabsorption of filtered urate in the proximal tubule is more complex, involving an interaction of two transport models. In the urate-anion exchange, in addition to URAT1, also the organic anion exchangers OAT4 and OAT10 participate, which are encoded, respectively, by the SLC22A11 and SLC22A13 genes29 (Fig. 1). In addition, in proximal tubules cells, the reabsorption of urate and monocarboxylate anions, such as lactate, is carried out by Na + -dependent cotransport. This process is mediated by the Na + -coupled monocarboxylate cotransporters SMCT1 and SMCT2 encoded, respectively, by the SLC5A8 and SLC5A12 genes30 (Fig. 1).
The exit of the UA towards the peritubular space is carried out by a basolateral transporter. In 2003, Jutabha et al. identified a new voltage-sensitive anion transporter (voltage-driven organic anion transporter 1), which facilitates the exit of urate from the cell.31 It is encoded by the SLC2A9 gene.32 Subsequently, it was named GLUT9 (Fig. 1), as it was known that it belongs to class II of the GLUT family (g lucose transporters) of hexose transporters (fructose, glucose).32 Studies of expression in Xenopus oocytes have shown that GLUT9 exchanges urate for glucose and fructose.33 It has been shown that GLUT9-mediated urate transport is voltage-dependent, which is an appropriate characteristic for urate efflux from the cell.32 Two different GLUT9 isoforms have been described ; in human kidneys, GLUT9L (540 amino acids) is located in the basolateral membrane of the cells of the proximal tubule, while the short isoform GLUT9S (511 amino acids) is located in the apical membrane of the collecting ducts.34
Mutations in the SLC2A9 gene are the cause of renal tubular hypouricemia type 2.28,35,36 GLUT9 is responsible for a large part of the reabsorption of urate, in such a way that the complete loss of its function leads to a massive excretion that is greater than that observed in renal tubular hypouricemia type 1, this is a reason why patients show such a low levels of UA (less than 0.5mg/dl) and values of fractional excretion greater than 150ml/100ml GFR.36 Heterozygous carriers have moderately reduced UA levels.36
Proximal Tubular Transporters Involved in Urate SecretionThe mechanisms involved in urate secretion by the proximal tubule are a mirror image of those involved in its reabsorption. In the basolateral region, urate transporters OAT1 and OAT3 move urate into the cell in exchange for α-ketoglutarate. The -αketoglutarate gradient is provided by the Na + cotransporter, NaDC3, encoded by the SLC13A3 gene (Fig. 2). Urate efflux into the lumen in the apical region is performed by ATP-stimulated transporters, ABCC4 (ATP Binding cassette Subfamily C Member 4; ATP-binding cassette subfamily C member 4, encoded by the ABCC4 gene) and ABCG2 (ATP Binding cassette Subfamily G Member 2; member 2 of the subfamily G of the ATP-binding cassette, encoded by the ABCG2 gene) and the apical electrogenic urate transporters NPT1 and NPT4, which are encoded, respectively, by the SLC17A1 and SLC17A3 genes (Fig. 2).30 It is noteworthy that it has been reported that the rs1165196 polymorphism of the above-mentioned sodium-dependent phosphate cotransporter 1 (NPT1) gene significantly reduces the risk of gout by increasing urate export towards the tubular lumen.37
Genetics and gout. Polymorphisms that favor hyperuricemia in the genes encoding the transporters URAT1, GLUT9, ABCG2 and ABCC4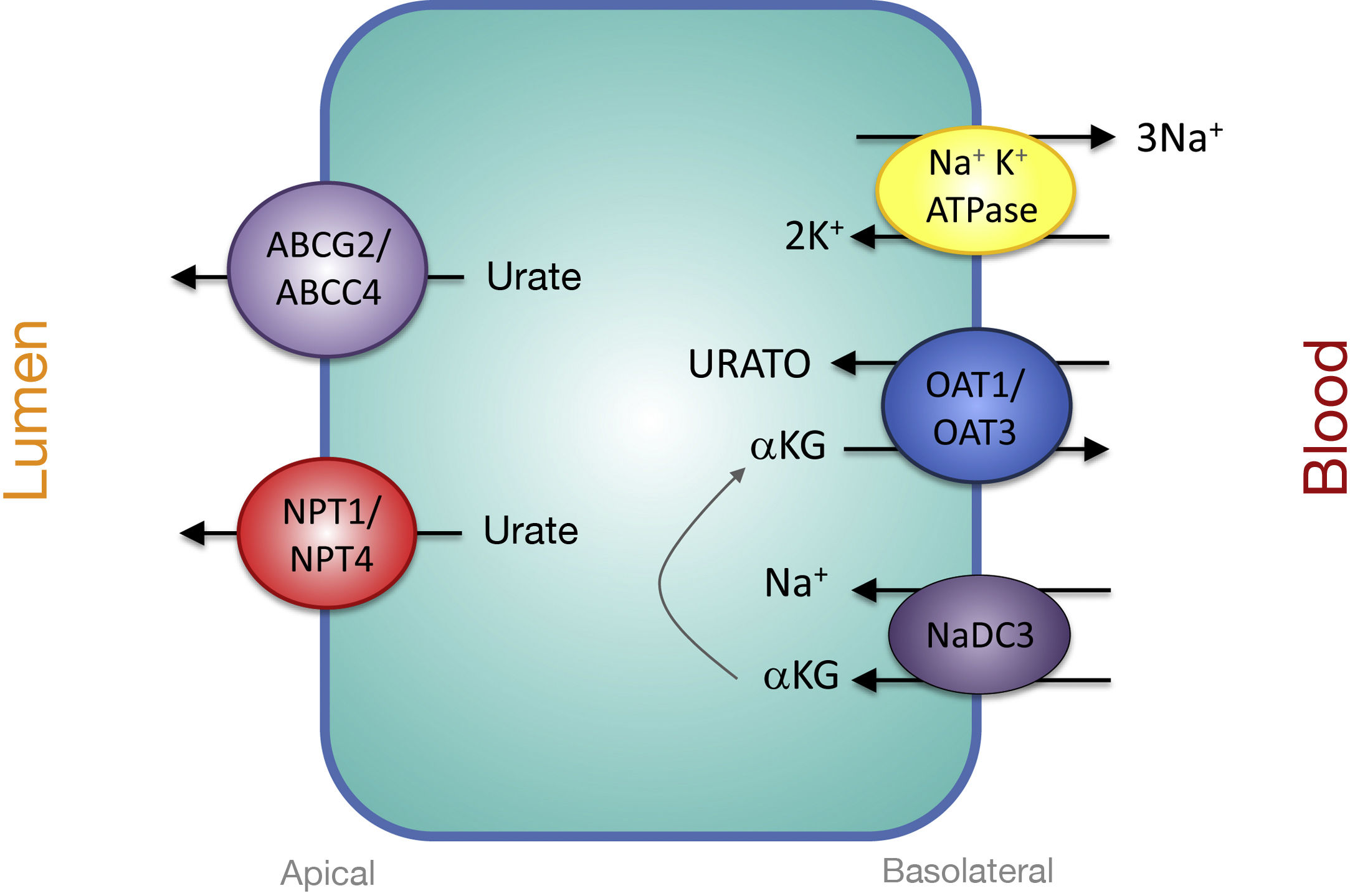
In 1992, Emmerson et al. attempted to demonstrate the existence of a genetic predisposition to gout by studying renal urate clearance in 37 pairs of normouricemic twins. The authors found that monozygotic twins had more similar urate clearance values than dizygotic twins.38 They calculated that the heritability of renal urate clearance was 60%, while the heritability of fractional excretion was 87%.
It has been shown that different polymorphisms in SLC22A12 can have an effect on the renal excretion of urate through an increase in the function of URAT1, which is why they have been associated with hyperuricemia and a decrease in its fractional excretion with the consequent risk of gout.39,40 In a study conducted in a cohort of 69 patients with gout, six different mutations were found in the gene SLC22A12 in 16 cases (23%).41
Genome-wide association methods have discovered common genetic variants of SLC2A9 associated with elevated serum urate levels and gout in human population cohorts associated to increased functional GLUT9 activity.42 Certain alleles of SLC2A9 have been related to tophaceous gout.43 This fact is remarkable because SLC2A9 is expressed in chondrocytes,44 which could be connected to the fact that some patients develop tophi and others do not.43
The secretory capacity of ABCG2 was confirmed by showing that the rs2231142 variant of ABCG2, which leads to the exchange of glutamine for lysine at codon 141, favors a 53% reduction in the rate of urate transport.45 Woodward et al. have estimated that at least 10% of all gout cases in Caucasians are attributable to the rs2231142 variant45; population studies have confirmed a link of UA levels and the presence of gout, although the association is stronger in men than in women.45 ABCG2 likely plays an important role in gout severity. Thus, a recent study found that a variant of ABCG2 is associated with a 50% increase in the frequency of tophaceous gout as compared with cases of the disease without tophi production.46 In 2012, Ichida et al. described that ABCG2 is expressed in extrarenal tissues. Thus, Abcg2 knockout mice show reduced intestinal excretion of urate.19 Ichida et al. have indicated that patients with mutations in the ABCG2 gene would be carriers of a "combined" type of hyperuricemia in which there would be an increase in the urinary excretion of UA (more than 600mg/day/1.73m2) together with a fractional excretion of UA less than 5.5ml/100ml GFR,19 which is difficult to understand from the renal point of view, unless there is associated glomerular hyperfiltration. Likewise, it has been shown in the Maori and Pacific populations of New Zealand that the ABCC4 variant rs4148500 is significantly associated with hyperuricemia and gout.47
Other genes related to gout caused by reduced renal excretion of uric acidGenome-wide association methods (GWAS) have made it possible to locate new loci related to gout associated with reduced renal excretion of UA, such as Nipa- Like domain containing 1 (NIPAL1)48 and Family with sequence similarity 35, member A (FAM35A).48,49NIPAL1 encodes a magnesium transporter; the functional analysis has not detected that it intervenes in the transport of urate, which suggests an indirect association with its renal management. FAM35A encodes a protein that plays an important role in the repair of DNA double-strand breaks. Localization analyzes in the human kidney have revealed that NIPAL1 and FAM35A are primarily expressed in the distal tubules, suggesting the involvement of the distal nephron in renal urate handling.48 At this point, it is convenient to remember autosomal dominant tubulointerstitial nephropathy (formerly known as juvenile familial hyperuricemic nephropathy) which is caused by mutations in the UMOD gene encoding the Tamm-Horsfall or uromodulin protein. Well, this protein is produced in the epithelial cells of the ascending branch of the loop of Henle. Does the absence of uromodulin in this nephropathy have repercussions by modifying urate transport by GLUT950 or does it intervene in the function of these “new” distal transporters?
EpilogueProgression to a clinically evident gout ranges from the presence of hyperuricemia to the deposition of monosodium urate crystals in various tissues with symptoms related to the innate immune response to these crystals and the expression of various genes that encode proteins that influence activation of the NLRP3 inflammasome.
Many genes are involved in renal and intestinal management as well as in the production of UA that are interrelated with certain factors such as age, sex and ethnicity and other modifiable factors such as diet and lifestyle.
The complexity of UA handling in the body means that it is not a "waste" product but rather a substance whose levels must be carefully guarded for its protective role as a powerful antioxidant, although a failure in the regulation system can contribute to the development of gout, kidney damage and high blood pressure. In this sense, the title of this work mentions that gout associated with reduced renal excretion of UA is a tubulopathy that we nephrologists do not treat. Certainly, it is a half truth, because patients appear at the nephrologist's office when they develop chronic kidney disease.
Conflict of interestsThe authors declare that they have no conflict of interest.
Please cite this article as: García-Nieto VM, Claverie-Martín F, Moraleda-Mesa T, Perdomo-Ramírez A, Tejera-Carreño P, Cordoba-Lanus E, et al. La gota asociada a reducción de la excreción renal de ácido úrico. Esa tubulopatía que no tratamos los nefrólogos. Nefrologia. 2022;42:273–279.



