



Phosphate balance is critical for life. Either acute or chronic hypo- or hyperphosphatemia may be harmful to animals and humans. Thus, mechanistic pathways exist to prevent the potential toxicity of phosphate disbalance. Phosphate intake absorption, the influx to and efflux from bone, and renal excretion maintain serum phosphate levels within the normal range. In mammals, different phosphatonins, such as fibroblast grow factor-23 (FGF23), and parathyroid hormone (PTH), are the two most commonly known molecules responsible for phosphate homeostasis through inhibiting renal phosphate reabsorption. FGF23 is an osteocyte derived protein with a critical role in phosphate and vitamin D homeostasis.1 Several studies have described the current understanding of FGF23 and the complications associated with its elevation.2 Primarily, FGF23 inhibits urinary phosphate reabsorption by reducing NPT2a and NPT2c transporters’ activity in proximal renal tubules while it also reduces calcitriol production.1,3 In the context of progressively declining kidney function, FGF23 increases adaptively to maintain normophosphatemia. However, this adaptive response becomes maladaptive since excessive circulating FGF23 may be associated with left ventricular hypertrophy (LVH),4 an increased risk of infections,5 endothelial dysfunction,6 inflammation,7,8 and a higher risk of death.9–11 Therefore, presently, FGF23 may not only be considered a biomarker of chronic kidney disease (CKD), but also a therapeutic target.2,12
In the current issue of Nefrologia, Vasquez-Rios and colleagues13 describe the clinical case of a 28-years-old female that received IV ferrous carboxymaltose (FCM) because of heavy uterine bleeding and iron deficiency anemia. The patient developed respiratory failure associated with severe hypophosphatemia (1–1.6mg/dl) due to renal phosphate wasting syndrome [fractional excretion of phosphate (FePi), and urine phosphate loss 3-fold above normal range]. Aggressive phosphate and vitamin D restitution were initiated, although serum phosphate did not improve until a few weeks later by the time symptoms disappeared. At discharge, approximately twelve weeks from the start of symptoms, the patient remained under oral calcitriol and oral supplementation of phosphate. The authors assessed c-FGF23 serum levels that were reported as within the normal range. However, they failed to report iFGF23, as neither was PTH, calcium, nor calcitriol serum levels.
How is FGF23 regulated?FGF23 can be measured in the circulation as two different isoforms, the full-length intact molecule (iFGF23) and the carboxy-terminal domain (cFGF23) that is mainly derived from proteolytic cleavage of iFGF23. cFGF23 domain interacts with the co-receptor α-Klotho, thereby enhancing FGF23-FGFR interaction.1,14 Several studies have shown the factors associated with the increase in circulating FGF23. Classic factors such as high serum phosphate,15 high calcium,16 and PTH disarrangements17 are not only partially responsible for FGF23 increment but also for adverse hard-outcomes in patients with CKD. However, some other factors may promote the increase in circulating FGF23 in the general population.
Both FGF23 and PTH regulate renal phosphate excretion. Thus, any medication that alters FGF23 and PTH homeostasis may be reflected in renal phosphate loss. FGF23-mediated phosphaturia upregulates the expression of FGFR1 in the kidney.18 Concomitantly, renal Klotho expression is reduced. Thus, phosphaturia may prolong hypophosphatemia via FGFR1 upregulation in the kidney.
PTH acts as a contra-regulatory hormone in mineral metabolism regulation. Indeed, PTH has been shown to increase FGF23 production directly and indirectly. On the contrary, increased FGF23 reduces PTH transcription. FCM seems to increase PTH, an effect that peaks at week five after administration.19 As PTH is not informed, we cannot elucidate whether PTH played a role in developing hypophosphatemia in this patient. However, we may hypothesize that PTH might have been critical for the long-lasting renal phosphate wasting given that maximum levels are expected after the fifth week of administration. It is unknown why the FCM administration overwhelms the contra-regulatory cross-talk between FGF23/Vitamin D/PTH. One possible explanation is that PTH upregulates to increase the efflux of phosphate and calcium from bone, given the persistent hypophosphatemia. Also, as phosphaturia decreases the renal expression of Klotho,18 it is unknown if parathyroid gland Klotho expression is also concomitantly reduced as seen in experimental models of CKD, in which the lack of the expression of Klotho over the parathyroid gland suppresses the inhibitory effect of FGF23 on the secretion of PTH.
Serum calcium, another regulator of FGF23, was not reported in this patient. Hypocalcemia inhibits FGF23 transcription to prevent FGF23-mediated calcitriol reduction.16 On the contrary, FGF23 regulates calcium homeostasis by inhibiting PTH transcription, vitamin D production, or blocking the transient receptor potential cation channel subfamily V (TPRV5) receptors in the gut and kidney. The use of FCM has been associated with transient calcium decrease.20 Together with FCM-mediated reduction in calcitriol, the drop in serum calcium may explain the increase in PTH, and thus transient hyperparathyroidism. However, the decline in serum calcium and the increase in serum PTH merely match in time. Therefore, another unknown mechanism may explain the later increase in serum PTH associated with FCM.
Vitamin D also regulates FGF23 homeostasis. FCM reduces calcitriol serum levels, which in turn may have to worsen hypophosphatemia.19 Since vitamin D serum levels were not analyzed, we cannot discard such an effect. It is known to what extent the initiation of calcitriol contributed to persistent hypophosphatemia by enhancing iFGF23 production.21 However, it is unlikely that calcitriol administration had contributed to iFGF23 increase since lower phosphate halts FGF23 transcription irrespective of calcitriol. Also, the nutritional deficiency of calcitriol could have been worsened by FCM.
The role of iron and inflammation in FGF23 regulationRecent evidence has demonstrated that iron deficiency and inflammation are critical regulators of FGF23 production and cleavage,7 irrespective of kidney function. Animal and human studies have shown that inflammation induces iron deficiency, thereby promoting Fgf23 transcription.7,19 Delineating the relationship between iron deficiency and FGF23 homeostasis may be beneficial for patients since either anemia, iron metabolism alterations, or inflammation are associated with a higher morbidity and mortality rate in the general and CKD population.
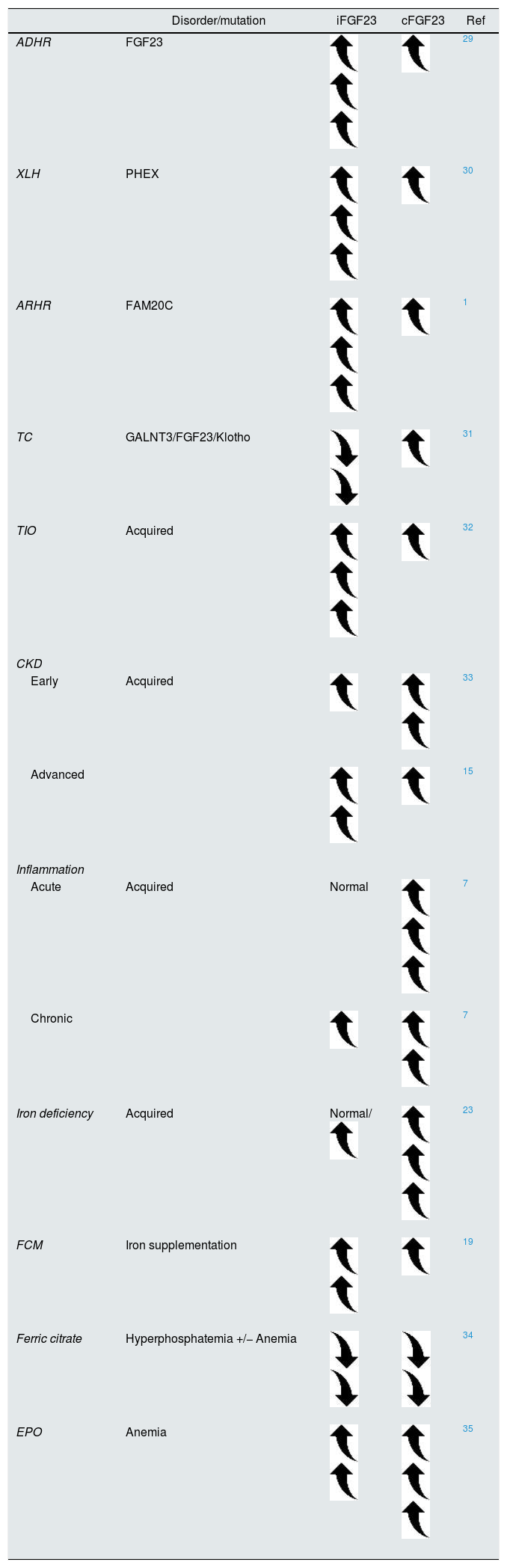
In healthy subjects, both FGF23 molecules remain within the normal range because of the tight balance between transcription, post-translational modifications, and cleavage. Different genetic or acquired disorders depict elevated iFGF23 compared to cFGF23. Autosomal dominant hypophosphatemic rickets (ADHR) is the prototype disease of iFGF23 excess encompassing clinically relevant hypophosphatemia, renal phosphate wasting, and reduced calcitriol levels. Similarly, early CKD patients also display similar clinical findings except for normal to high phosphate serum levels. On the contrary, iron deficiency is likely to change the iFGF23-to-cFGF23 ratio to increase cleavage, whereas iFGF23 continues at most normal or slightly elevated (Table 1). Iron supplementation restores the iFGF23-to-cFGF23 ratio with a transient increase in iFGF23, mimicking the ADHR phenotype (Table 1).
Approximate representation of FGF23 homeostasis across different clinical settings.
| Disorder/mutation | iFGF23 | cFGF23 | Ref | |
|---|---|---|---|---|
| ADHR | FGF23 | 29 | ||
| XLH | PHEX | 30 | ||
| ARHR | FAM20C | 1 | ||
| TC | GALNT3/FGF23/Klotho | 31 | ||
| TIO | Acquired | 32 | ||
| CKD | ||||
| Early | Acquired | 33 | ||
| Advanced | 15 | |||
| Inflammation | ||||
| Acute | Acquired | Normal | 7 | |
| Chronic | 7 | |||
| Iron deficiency | Acquired | Normal/ | 23 | |
| FCM | Iron supplementation | 19 | ||
| Ferric citrate | Hyperphosphatemia +/− Anemia | 34 | ||
| EPO | Anemia | 35 | ||


ADHR, autosomal dominant hypophosphatemic rickets; XLH, X-linked hypophosphatemic rickets; ARHR, autosomal recessive hypophosphatemic rickets; TC, tumoral calcinosis; TIO, tumor induced-osteomalacia; FAM20C, the family with sequence similarity 20; CKD, chronic kidney disease; FCM, ferric carboxymaltose; EPO, erythropoietin.
The results showed by Vasquez-Rios et al.13 are consistent with those reported by Wolf and colleagues,19 in which adults with iron deficiency-associated anemia were submitted to receive either ferric carboxymaltose or ferumoxytol. Iron stores and hemoglobin levels were restored in both groups. However, more importantly, together with a remarkable drop in serum phosphate, iFGF23 transiently raised (peaked by week two) in those assigned to FCM compared to the ferumoxytol group.19 In parallel, with the increase in iFGF23, FePi also rose. Simultaneously, cFGF23 decreased to the normal range. More interestingly, 50% of the patients that received FCM showed severe hypophosphatemia (serum phosphate <2mg/dl), whereas nearly 10% displayed extremely low serum phosphate <1.3mg/dl. The authors recognized that the effect of FCM on phosphate homeostasis is magnified in non-CKD compared to CKD patients, perhaps because CKD subjects may be protected due to chronic phosphate overload and the renal resistance to the action of FGF23. Indeed, a normal glomerular filtration rate (GFR) was identified as an independent predictor for hypophosphatemia so that the better the GFR, the higher the risk for incident hypophosphatemia.
Inflammation has different roles in mineral and iron metabolism. Various studies have demonstrated the self-perpetuating relationship between inflammation and FGF23 production. In an experimental study, David V et al.7 showed that inflammation induces a decrease in serum iron, an increase in ferritin, and an increment in circulating cFGF23 and osseous expression of Fgf23 mRNA. Interestingly, acute or chronic Inflammation may have different effects on FGF23 transcription (Table 1). While in the acute setting, iFGF23 remains normal, cFGF23 increases dramatically. On the contrary, chronic inflammation increases both molecules, although cFGF23 increases to a greater extent than iFGF23.7 Systemic inflammation triggers the other two different pathways to induce FGF23 production. One is the calcineurin/nuclear factor of activated T cells (NFAT) pathway. The other involves the nuclear factor κβ (NFκβ), the tumor necrosis factor-alpha (TNF-α), interleukin 6 (IL6), and IL-1β that also promote FGF23 transcription14 (Fig. 1). In this line, inflammation mediated by IL-1β increases FGF23 transcription irrespective of the iron status,7 given the direct upregulation of inflammation over the hypoxia-inducible factor 1α (HIF1α).
 EPO production together with BM erythroid cells apoptosis (BM pro-E cells) and a reduction in colony-forming for erythroid progenitors (BFU-E colonies). Inflammation and iron deficiency activate HIF-1α. Hepcidin also stimulates FGF23 transcription independently of the inflammatory milieu. Moreover, Inflammation itself downregulates bone expression of PHEX and DMP1. CKD is usually accompanied by iron deficiency, reduce EPO production, inflammation, and hepcidin increase. *Green lines: upregulation; red lines: downregulation.")
Schematic view of the relationship between Inflammation, iron, and FGF23. Bidirectional pathways have been described between Inflammation, iron metabolism, and FGF23 production. CKD is one of the leading causes of secondary FGF23 excess. CKD increases Inflammation, which, in turn, promotes FGF23 transcription. FGF23 targets hepatocytes to induce cytokine production, which ends in a vicious circle. Inflammation increases the secretion of hepcidin by the liver, thereby degrading ferropontin and boosting iron retention in enterocytes and macrophages. Iron deficiency upregulates hepcidin, which may also promote FGF23 transcription. The resultant iron deficiency promotes FGF23 production and cleavage. FGF23, directly and indirectly, reduce renal and bone marrow (BM) EPO production together with BM erythroid cells apoptosis (BM pro-E cells) and a reduction in colony-forming for erythroid progenitors (BFU-E colonies). Inflammation and iron deficiency activate HIF-1α. Hepcidin also stimulates FGF23 transcription independently of the inflammatory milieu. Moreover, Inflammation itself downregulates bone expression of PHEX and DMP1. CKD is usually accompanied by iron deficiency, reduce EPO production, inflammation, and hepcidin increase. *Green lines: upregulation; red lines: downregulation.
Inflammation and iron deficiency partially share the pathways by which FGF23 transcription is upregulated. Mechanistically, HIF-1α has been implicated as one of the vital regulators of this relationship. HIF-1α regulates oxygen detection and adaptation of cells to hypoxia. Physiologically, tissue hypoxia activates target response genes to promote erythropoietin (EPO) production, EPO sensitivity, iron absorption, iron transport, and heme synthesis. Iron regulates the activation and degradation of HIF-1α. As such, iron overload promotes the degradation of HIF-1α, whereas iron deficiency stabilizes HIF-1α through the inhibition of prolyl hydroxylase activity, meaning that HIF-1α is not hydroxylated nor degraded. Hence, iron deficiency inhibits HIF-1α degradation, so FGF23 is produced.
On the contrary, HIF-1α inhibition attenuates the contribution of inflammation on FGF23 transcription. Some of these mechanisms may be perpetuated because inflammation-associated functional iron deficiency promotes the upregulation of hepcidin by the liver,7 which reduces gastrointestinal iron absorption and its release from the reticuloendothelial system (Fig. 1).22 True iron deficiency also upregulates FGF23 cleavage.20 Indeed, women with heavy uterine bleeding, iron-deficiency anemia, and normal renal function show at most normal iFGF23 serum levels, whereas cFG23 are markedly increased,20 highlighting the role of iron deficiency in FGF23 transcription and cleavage23 (Table 1). Therefore, there exist similarities in the response of FGF23 to inflammation and iron deficiency that take this relationship into the spotlight for future research. Although markers of inflammation in the patient mentioned above were not reported, iron-deficiency-mediated overt inflammation may have been implicated in the increment of FGF23 and the clinical outcome.
Some other possibilities for the effect of iron supplementation on the increase of FGF23 remain under study. Several authors have suggested that some iron preparations, such as FCM, restore iron deposits, and reduce the degradation of FGF23 within osteocytes by enhancing O-glycosylation by polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3).1 Another possibility is that certain iron preparations promote the ectopic production of FGF23 beyond the bone. In this regard, different iron preparations have been associated with the production of some inflammatory cytokines and oxidative stress.24,25 Specifically, iron prescriptions, including FCM, increase the percent of mononuclear cells with reactive oxygen species and intercellular adhesion molecule-1 (ICAM-1), which are keen on secreting TNF-α and IL-6 that could stimulate FGF23 transcription.26
Other factors regulating FGF23Finally, other factors regulating FGF23 transcription seem unlikely related to hypophosphatemia development in this case report. By activating the FGFR1/α-Klotho/ERK pathway, FGF23 increases sodium reabsorption through sodium/chloride cotransporter in distal tubules. Hence, FGF23 promotes volume overload and high blood pressure.2,27 It is unknown whether FGF23-mediated volume overload contributed to the FGF23 increment and respiratory failure in this patient. Erythropoietin (EPO) also shows a two-way relationship with FGF23. Indeed, increased FGF23 reduces bone marrow EPO expression (Fig. 1). On the contrary, EPO administration promotes FGF23 transcription.2 The administration of EPO in this patient is not informed, although it is remote as CKD was not apparent. The reduction of FGF23 in experimental models of CKD rescues renal anemia by increasing EPO production and response and decreases inflammation, which is one of the determinants of anemia development, FGF23 production, and death.28
SummaryThe findings described by Vasquez-Rios and colleagues13 demonstrate the complex network between iron deficiency anemia, inflammation, and mineral metabolism alterations. Given that some of the observed disturbances displayed following the use of FCM are not completely understood, further research is warranted. It is difficult to determine to what extent each of the variables independently contributed to the FGF23 increase. Other mechanisms by which FCM promotes FGF23 transcription should be explored. In the meantime, serum and urinary phosphate and vitamin D monitoring and perhaps supplementation should be thoroughly considered in those patients with conserved renal function submitted to receive FCM. Hopefully, in the forthcoming years, we will learn much more about FGF23 not as a phosphate regulator or a biomarker but as a potential therapeutic target.2
FundingThis study was supported by grants from the National Institute of Health Carlos III (FIS 18/0138) and the Consejeria de Salud of Junta de Andalucía (PI-0169-2020). JR Muñoz-Castañeda is a senior researcher supported by the Nicolas Monardes Programme from Consejeria de Salud-SAS (Junta de Andalucía). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Conflict of interestAMM has received lecture fees from Fresenius Medical Care, Medtronic, Viphor, and Astrazeneca in the last two years. All the other authors declare no conflict of interest to disclose. The companies pointed in the financial disclosures had no role in the study design, collection, analysis, interpretation of data nor preparation of the manuscript.



