



Kidney problems are among the most common complications in sickle cell disease (SCD). They occur early in childhood and are one of the main factors related to mortality in these patients. The main underlying pathogenic mechanisms are vaso-occlusion and haemolysis. The renal medulla has ideal conditions for the sickling of red cells due to its low partial pressure of oxygen, high osmolarity and acidic pH. Initially, sickle-cell formation in the vasa recta of the renal medulla causes hyposthenuria. This is universal and appears in early childhood. Microscopic and macroscopic haematuria also occur, in part related to renal papillary necrosis when the infarcts are extensive. Release of prostaglandins in the renal medulla due to ischaemia leads to an increase in the glomerular filtration rate (GFR). Adaptively, sodium reabsorption in the proximal tubule increases, accompanied by increased creatinine secretion. Therefore, the GFR estimated from creatinine may be overestimated. Focal segmental glomerulosclerosis is the most common glomerular disease. Albuminuria is very common and reduction has been found in 72.8% of subjects treated with ACE inhibitors or ARB. Recent evidence suggests that free haemoglobin has harmful effects on podocytes, and may be a mechanism involved in impaired kidney function in these patients. These effects need to be better studied in SCD, as they could provide a therapeutic alternative in sickle cell nephropathy.
Las complicaciones renales se encuentran entre las más frecuentes de la enfermedad falciforme (EF), aparecen tempranamente desde la infancia y constituyen uno de los principales factores relacionados con la mortalidad en estos pacientes. La vasooclusión y la hemólisis son los principales mecanismos patogénicos subyacentes. La médula renal reúne condiciones ideales para la falciformación de los hematíes debido a su baja presión parcial de oxígeno, elevada osmolaridad, y pH ácido. Inicialmente, la falciformación en los vasa recta de la médula renal es la causa de hipostenuria, que es universal y aparece en la infancia temprana. Existe también hematuria, microscópica y macroscópica en parte relacionada con la necrosis papilar renal cuando los infartos son extensos. La liberación en la médula renal de prostaglandinas debido a la isquemia se relaciona con el aumento del filtrado glomerular (FG). De forma adaptativa, aumenta la reabsorción de sodio en el túbulo proximal, que se acompaña de un aumento de la secreción de creatinina. Por ello, el FG estimado a partir de la creatinina puede estar sobreestimado. La glomeruloesclerosis focal y segmentaria es la glomerulopatía más común. La albuminuria es muy frecuente y se ha observado reducción en el 72,8% de los sujetos tratados con IECAs o ARA-II. Recientes evidencias sugieren que la hemoglobina libre tiene efectos nocivos sobre los podocitos, pudiendo ser un mecanismo implicado en la alteración de función renal que presentan estos enfermos. Estos efectos han de ser mejor estudiados en la EF, ya que podrían constituir una alternativa terapéutica en la nefropatía falciforme.
Key concepts
- •
Kidney complications are common in patients with sickle cell disease (SCD).
- •
Vascular occlusion and hemolysis are the main pathogenetic mechanisms of sickle cell nephropathy.
- •
It often presents as hyposthenuria, hematuria, albuminuria, increase in glomerular filtration rate and in the function of proximal tubules, acute kidney failure, or chronic kidney disease.
- •
Treatment with ACEIs or ARBs reduces albuminuria in most subjects.
- •
Free hemoglobin has deleterious effects on podocytes, although there are no studies that have validated this observation in patients with SCD.
- •
Despite the advances made in recent years, it is necessary to continue studying the pathophysiology of FD and its relationship with kidney damage, the fact is that there are no effective pharmacological treatments that prevent kidney damage and its progression.
Sickle cell nephropathy is the term used to refer to the set of manifestations of sickle cell disease (SCD) in the kidney. Kidney complications are one of the most frequent and serious in SCD. They are often present since childhood, and are one of the leading causes of death in adults.1
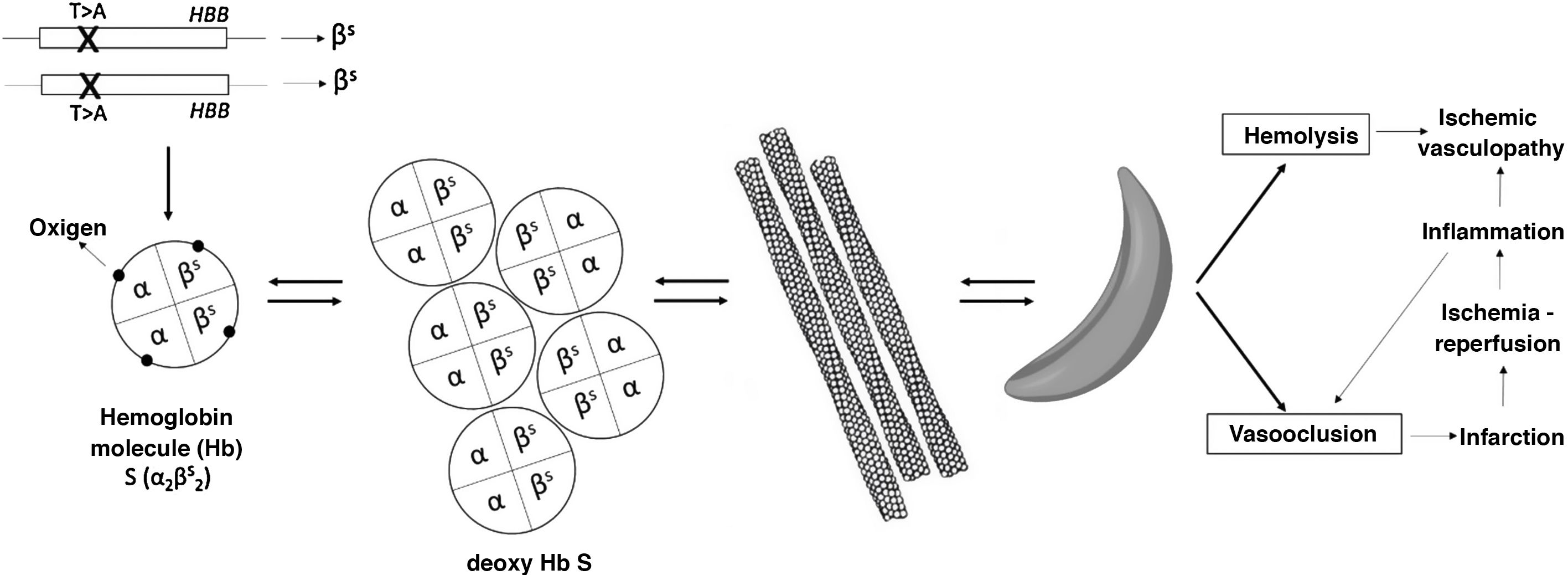
SCD (or sickle cell disease) encompasses the set of phenotypes resulting from the combination of the mutation responsible for hemoglobinopathy S, in one of the alleles of the beta globin (HBB) gene, with another mutation in the other allele. Thus, the most serious variants are the homozygous state (S/S), to which the term “sickle cell anemia” refers, and the combination with a mutation that causes a silent allele (S/β°-thalassemia). Other forms are the double heterozygosity S/C or S/β+-thalassemia. When the mutation that causes hemoglobinopathy S coexists with a normal allele, that is, the state of carrier, it is called sickle cell trait, which is not a disease and is not included in the definition of SCDF. In sickle cell trait, around 40% of the hemoglobin (Hb) is Hb S. Since Hb is a tetramer made up of 2 alpha globins and 2 beta globins, alterations in the beta globin gene can coexist with mutations that reduce the production of alpha globin (α-thalassemias).2
Hb S is poorly soluble when deoxygenated and gives rise to rod-shaped polymers that give the red blood cell the characteristic sickle (in Latin, falx, falcis) or crescent shape (Fig. 1). This causes hemolysis and vasoocclusion, the main pathogenic phenomena of SCD, leading to endothelial dysfunction and vasculopathy, ischemia/reperfusion damage, oxidative stress, hypercoagulability, nitric oxide deficiency (free Hb resulting from intravascular hemolysis binds to nitric oxide and sequesters it), platelet activation, and increased neutrophil adhesiveness. All of this gives rise to acute and chronic manifestations that can potentially affect any organ. The most frequent are chronic anemia and painful bone crises. Other manifestations are functional asplenia/hypoasplenia, sepsis, silent cerebral infarcts and stroke, retinopathy, pulmonary hypertension, pulmonary fibrosis, acute chest syndrome, avascular necrosis of the hip or shoulder, ulcers in the lower extremities, priapism, or sickle cell nephropathy itself.2,3
Sickle cell trait confers partial protection against malaria, which justifies that SCD predominantly affects people of African origin, although it is also prevalent in individuals from the Middle East, India, and South and Central America.4 In sub-Saharian Africa, the prevalence of sickle cell trait ranges between 10% and 30%, and in some countries the disease affects 2% of the population.5 The increase in the migratory flow from endemic areas, the establishment of universal neonatal screening programs in the autonomous communities and the improvement in survival are responsible for the increase in the number of patients being followed in our country. However, there are still no data on the real prevalence of the disease in Spain. According to data published at the beginning of 2019,6 826 patients with SCD were included in the Spanish Registry of Hemoglobinopathies (REHem), a non-population registry sponsored by the Spanish Society of Pediatrics (SEHOP) and with the participation of some centers. Most of the individuals included were concentrated in the Community of Madrid, Catalonia and, to a lesser extent, in the Valencian Community. Most of the children (63.3%) were born in Spain, most of whom (51.4%) were diagnosed through neonatal screening. In Madrid, the incidence of SCD is 0.16 per 1,000 births.6
SCD should be suspected in the presence of hemolytic anemia or other clinical manifestations of the disease, and an with an ethnic origin of risk. The clinical manifestations will not appear, even in the most severe forms, until past few months after birth, as the fetal Hb (Hb F) decreases throughout the first year of life.
Ethnicity is not always a marker. Anemia and hemolysis traits may be absent in some cases in certain genotypes. We will generally observe sickle red cells in the peripheral blood smear, although they are rare in the Hb S/C form, in which dianocytes cells predominate. Mean corpuscular volume (MCV) may decrease (microcytosis) in the Hb S/C form or in the forms associated with β-thalassemia (Hb S/β0-thalassemia or Hb S/β+-thalassemia) or α-thalassemia. Generally, the diagnosis will be completed with a study of Hb variants using high performance liquid chromatography (HPLC) and capillary electrophoresis. In certain cases, will be useful a confirmatory genetic study.7,8
Allogenic hematopoietic stem cell transplantation (HSCT) is the only curative treatment available. However, its application in our environment is limited by the lack of suitable donors and the associated morbidity, especially when it is performed beyond adolescence. In the United States, less than 15% of patients have an HLA-identical family donor.9 Treatments based on gene editing are under study, and the results are promising. Hydroxyurea is still the only drug approved in Europe to treat the disease. This drug induces the production of Hb F, which inhibits the polymerization of Hb S and therefore, the sickle formation. However, the Hb F levels obtained with the maximum tolerated dose are usually between 20 and 30%, so Hb S continues to represent the predominant Hb.10 Therefore the benefit of hydroxyurea is partial. Furthermore, there is insufficient evidence about its benefit in patients with non-Hb S/S or Hb S/β°-thalassemia genotypes or about the prevention of chronic complications.11 In addition, the effect of different drugs already approved by the FDA, such as L-glutamine, crizanlizumab and voxelotor, on the chronic complications of SCD is still uncertain. Thus, the life expectancy of patients with SCD in developed countries is still reduced by 20 or 30 years.9 In this context, this review aims to constitute a tool for the care of patients with sickle cell nephropathy.
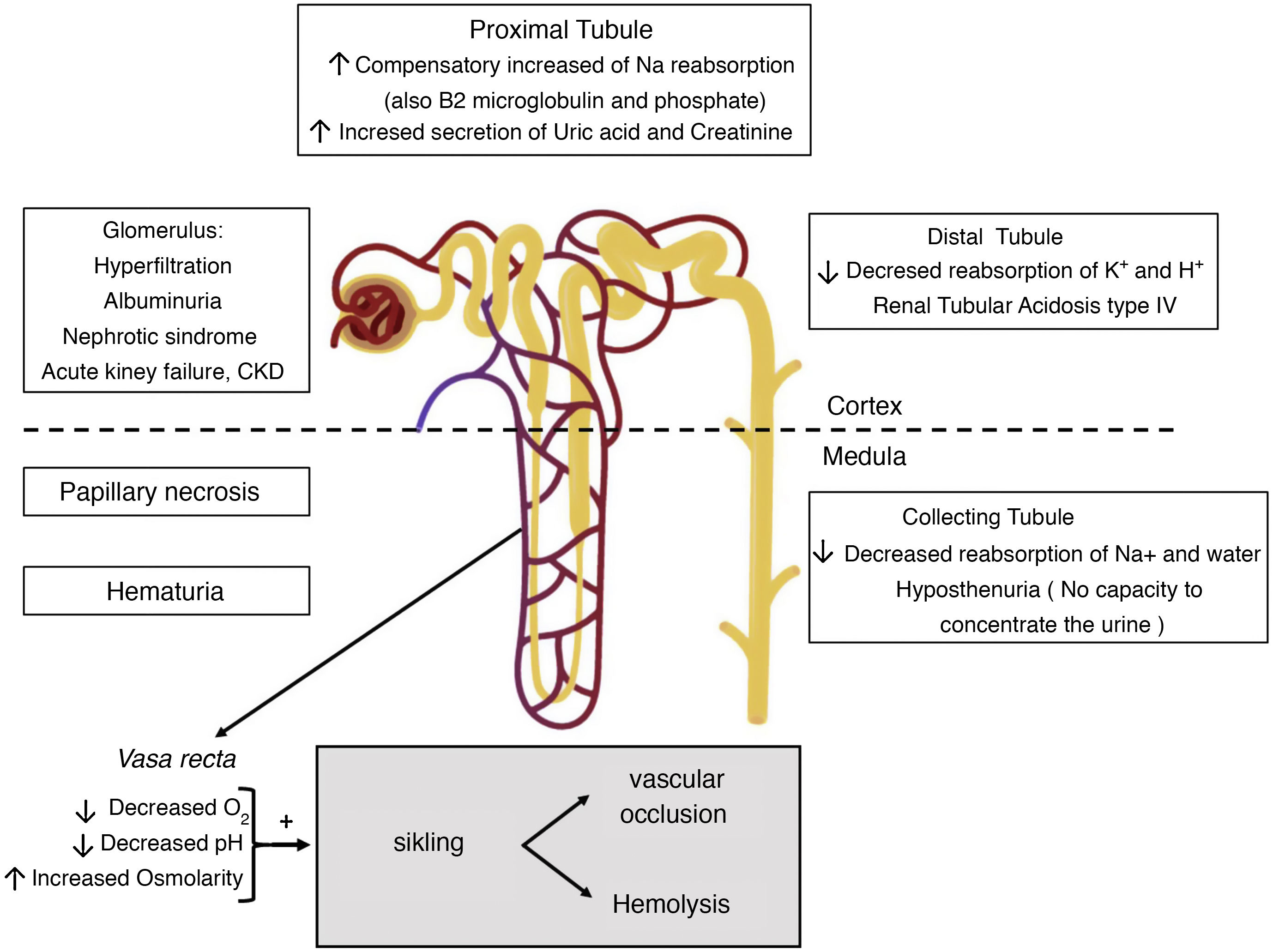
Clinical manifestationsHyposthenuriaHyposthenuria is defined as the inability to concentrate urine more than 450mOsm/kg under conditions of water deprivation, and it is a universal finding in SCD.12 Appears in early childhood and is irreversible after the 15 years of age in individuals with genotype S/S or S/β°-thalassemia.13 in individuals with sickle cell trait hypostenuria is observed around the age of 10.14
The environment of the renal medulla, which is critical for the reabsorption of water and solutes, is also ideal for sickling (Fig. 2). His partial pressure of oxygen of 35−40 mmHg is below the threshold for sickling (45 mmHg); the high osmolarity dehydrates red blood cells and concentrates Hb S; and its acidic pH decreases the affinity of Hb for oxygen (Bohr effect).15 For this reason, sickle formation initially occurs in the vasa rectus of the medulla, the result is that the sodium reabsorbed by the collecting tubule cannot be washed away by the circulation, preventing the reabsorption of free water and leading to medullary congestion. With time, repeated cycles of ischemia and infarction lead to destruction of the vasa recta, the formation of collaterals, and loss of the longest loops of Henle that descend into the deep medulla and the papilla and are originated from the juxtamedullary nephrons. However, the ability of urinary dilution remains intact as it depends essentially of cortical nephrons that have shorter loops of Henle and do not reach beyond the outer medulla and are irrigated by peritubular capillaries.13,15
Hyposthenuria is often manifested as nocturnal enuresis, polyuria, and increased risk of dehydration in case of insufficient water intake or increased extrarenal losses, favoring the appearance of vaso-occlusive crises. For this reason, a liquid intake of 3−4 l/day is recommended in patients with sickle cell disease.7 The degree of hyposthenuria is proportional to the concentration of Hb S and, in young children, treatment with hydroxyurea for 24 months was associated with greater urinary concentrating capacity compared to treatment with placebo.16
Tubular acidosisPatients with SCD may have a defect in the distal secretion of hydrogen ions and potassium, leading to incomplete type IV renal tubular acidosis (distal renal tubular acidosis with hyperkalemia), which accompanies hyposthenuria. Although the exact pathogenic mechanism is unknown, it may be due to a reduction in the electrochemical gradient in the collecting ducts, due to impaired medullary blood flow resulting in hypoxia. As the main damage occurs in the deepest segment of the loop of Henle, it is very rare to have a severe defect of urinary acidification and therefore the acidosis is incomplete. Hyperkalemia is rare and mild, unless there are other circumstances that affect the renal compensatory mechanisms, such as the administration of drugs that block the renin-angiotensin-aldosterone system (angiotensin converting enzyme inhibitors [ACEI] or antagonists of the angiotensin II [ARA-II]), potassium-sparing diuretics, or beta-blockers, excessive potassium intake, rhabdomyolysis, or kidney failure.15
Hematuria and renal papillary necrosisHematuria is common in patients with SCD, and its prevalence could be approximately double in people with sickle cell trait.17 It may occur at any age. It can be microscopic or, more often, macroscopic and self-constrained. It is due to vasoocclusive events and microinfarctions affecting the renal papillae. It is frequently unilateral coming from the left kidney, presumably for anatomical reasons: the left renal vein is longer and is compressed between the aorta and the superior mesenteric artery (nutcracker phenomenon), so it is subjected to greater venous pressure that increases relative hypoxia in the renal medulla, which favors sickling.12,15 Occasionally, these infarctions may be extensive, causing renal papillary necrosis (RPN), the prevalence of which is estimated in 30–40% of homozygous patients (Hb S/S). Its clinical presentation goes from asymptomatic hematuria (not always present) to acute pain, fever and acute obstructive renal failure even with hydronephrosis.15
The RPN can be diagnosed by renal ultrasound, although computed tomography (CT) urography is more sensitive and may be necessary to confirm the diagnosis,7 always using prophylactic measures such as volume expansion against nephrotoxicity by iodinated contras. On ultrasound, the earliest finding of RPN is an increased echogenicity of the medullary pyramids (the innermost medullary area), which in the absence of hypercalciuria in a patient with SCD and hematuria suggests RPN. Thereafter it can be seen calcification of medullary pyramids with a typical pattern of shadow “in the form of garland” surrounding the renal pelvis or defects of echogenicity in any pyramid due to the loss of the papilla.15
In cases of gross hematuria, it may be advisable to maintain a forced diuresis at 4 l/1.73 m2 by hydration, preferably with hypotonic sera, and the use of thiazide or loop diuretics. This measure will decrease osmolarity in the medulla and could reduce sickle formation in the vasa rectus, and help to clear clots from the urinary tract. Volume expansion with saline should be avoided, as it would be ineffective in reducing plasma osmolarity. Alkalinization of the urine could potentially be useful to increase the affinity of Hb for O2 and reduce sickness, and tubular toxicity of hemoglobinuria. Bed rest may also be appropriate to avoid detachment of the microthrombi.15
As in other serious complications, or those that do not respond to the treatments, the transfusion of red blood cells or erythrocyte exchange, will rapidly decreases the proportion of Hb S. This option must be carefully evaluated, since it does not free of risks. Simple transfusion may also be indicated if bleeding has been significant. Desmopressin has been used successfully in some patients with severe or refractory hematuria. This molecule could act at various levels: (1) decreasing plasma osmolarity, thus increasing the hydration of the red blood cells and decreasing the concentration of Hb S and its sickle formation or (2) promoting coagulation by increasing the levels of factor VIII and factor of von Willebrand up to supraphysiological levels.18 In this line, where other measures have failed, it is also described the use of antifibrinolytic agents, such as tranexamic acid or aminocaproic-ɛ acid, at a low dose and with care because of the risk of thrombosis.19 In isolated cases may be required selective embolization of the affected renal segment.20 Unilateral nephrectomy is not recommended, due to the risk of recurrence in the other kidney. In cases of persistent hematuria, iron replacement may be necessary.
HyperfiltrationHyperfiltration or increased glomerular filtration rate (GFR) (>130 ml/min/1.73 m2 in women and >140 ml/min/1.73 m2 in men) typically precedes albuminuria in patients with SCD, and may be present since early childhood. In a study in homozygous adult patients, about half of the subjects under 40 had hyperfiltration and in half of them it was associated with albuminuria.21
Renal plasma flow is increased, even more than GFR, so that the filtration fraction is decreased in patients with SCD compared to healthy subjects. This fact could reflect the selective loss of juxtamedullary nephrons, which present a higher filtration fraction than cortical nephrons. Renal hyperperfusion is not explained by the increase in cardiac flow due to anemia, since it is not reversed by repeated transfusions of red blood cells. Moreover, other conditions associated with chronic anemia do not usually cause renal hyperperfusion. It is probably related to the release in the renal medulla of vasodilator substances, such as prostaglandins, as a result of ischemia. The vasodilatation will reduce renal vascular resistance and increase renal plas flow. Inhibition of prostaglandin synthesis with indomethacin has been associated with a reduction in GFR. In this manner, the kidney exemplifies, in a single organ, the “perfusion paradox” that occurs in SCD: hypoperfusion is prevalent in the microvascular beds due to vaso-occlusion by Hb S, whereas hyperperfusion characterizes the systemic circulation and some regional circuits. The role of nitric oxide (NO) in this process is not known. However, as a result of intravascular hemolysis, Hb and heme group are released into the circulation, which chelate NO, explaining the defect or resistance to NO that characterizes SCD. In SCD it has has been observed an increase in the renal expression of the enzyme heme oxygenase-1 (HO-1). HO-1 catabolizes the heme group that accumulates in the kidney as a consequence of intravascular hemolysis, reducing oxidative stress and releasing carbon monoxide (CO), which has vasodilatory properties and could counteract the vasoconstriction induced by the heme group.13,22
Finally, it is important to note that hyperfiltration in SCD is not only associated with hemodynamic changes, but it is also due to an increase the glomerular filtration coefficient (Kf). Kf is the product of the total filtrate surface and the hydraulic permeability of the glomerular capillary wall; its increase could reflect at least an increase in the filtration surface due to glomerular hypertrophy, which is a feature of SCD and which, in models of the disease, accompanies an increase in glomerular plasma flow and intraglomerular pressure.13
Increased proximal tubular functionThe increase in GFR is related to an adaptive increase in proximal tubular function as an increase in sodium reabsorption. Also, it has been described an increased reabsorption of β2-microglobulin and phosphate (hyperphosphataemia may exist, especially if phosphate overload occurs, as in hemolysis patients), as well as increased secretion of uric acid and creatinine, and an increase in maximal transport of para-amino hippuric acid.13 The, increased reabsorption of sodium requires oxygen consumption and favors tubular damage due to increased oxidative stress. Furthermore, increased oxygen consumption will exacerbate hypoxia and promote sickness, aggravating kidney damage.13
ProteinuriaHemodynamic changes previously described may cause damage in the glomerular endothelium and podocytes, thus causing proteinuria,14,23 which subsequently causes tubulointerstitial damage.13 Podocyte injury leads to focal adhesions to the parietal epithelium.24 These adhesions cause focal and segmental glomerulosclerosis, which is the most common glomerulopathy in SCD, reported in up to 39% of kidney biopsies performed in patients with SCD and proteinuria and/or kidney failure. Other histological patterns such as membranoproliferative glomerulonephritis and thrombotic microangiopathy are less common. Hypertrophy of the glomeruli and hemosiderin deposits in the tubular cells are almost universal findings.25
Albuminuria is present in up to 27% of patients in the first 3 decades of life, and in up to 68% in older patients.26 However, the presence of proteinuria in the nephrotic range is rare and occurs in only 4% of patients with SCD.27 One study carried out over 5 years in 98 patients showed that albuminuria > 500 mg/g Cr was associated with progression to chronic kidney disease (CKD).28
It is recommended to start treatment with ACEI or ARA-II in SCD children and adults with albuminuria >100 mg/mmol (884 mg/g).12 The effect of this treatment has been evaluated in a randomized clinical trial with 22 patients and in another 7 observational studies including a total of 114 patients. A reduction in albuminuria was observed in 72.8% of treated subjects; most of the subjects had severe albuminuria. However, the effect of these drugs on the prevention of CKD is unclear. The dose should be adjusted if the GFR is <45 ml/min/1.73 m2. Given that patients with SCD have an increased risk of developing hyperkalemia, the GFR and serum potassium should be controlled after a week of treatment and before increasing the dose, and should be suspended during acute processes or before surgical interventions, administration of radiocontrasts or endoscopy preparations.29 As many patients have low or normal blood pressure, they can cause postural hypotension, so administration of the medication at bedtime may be beneficial.12
Blood pressure reductionIn 2014, a meta-analysis concluded that individuals with the Hb S/S genotype have significantly lower diastolic, systolic and mean blood pressure than healthy controls with the same age and sex (−8.37, −2.32 and −8, 41 mmHg, respectively).29 This fact could be related to the defect in urinary concentration. In patients with sickle cell anemia, albuminuria ≥300 mg/g creatinine and renal failure seem to be associated with resistance to the development of hypertension (HTN).30 A study including 163 patients suggests that a systolic blood pressure (SBP) between 120 and 139 mmHg or a diastolic blood pressure (DBP) between 70 and 89 mmHg define a category of relative hypertension in SCD, which is associated with an increased risk of pulmonary hypertension (PH) and renal dysfunction.31 Based on the benefits observed in the general population, it is recommended to maintain BP ≤ 130/80 mmHg.29
Acute kidney failureAcute renal failure occurs between 4–10% of patients admitted with SCD, being more frequent in those patients with acute chest syndrome (13.6% if it is severe and associated with pulmonary hypertension, which suggests venous congestion) than in patients with acute episodes of bone pain (2.3%).32,33 During episode of pain associated vasooclusive crises it has been observed a reduction of 15% in the creatinine clearance that is reversible.34 Volume depletion, rhabdomyolysis, infections, or the use of non-steroidal analgesics predispose to acute renal failure.13 Likewise, iron overload often occurs in SCD and requires chelating therapy; the most widely used agent is deferasirox. This drug may produce a reversible increase in serum creatinine in a dose-dependent manner, therefore, in the event of small increases in creatinine, a dose reduction should be considered.
Chronic kidney diseaseIn a study carried out in 410 patients between the ages of 2 and 21 with SCD, 26.5% had CKD stage 1, 14.5% and 11.6% had stage 2.35 In another study, 10.5% of deaths in adult patients with SCD were due to CKD.36 In homozygous patients older than 60 years, CKD was the main contributing factor to mortality in 43% of cases.37
Hydroxyurea, the only drug approved in our country for SCD, is metabolized in the kidney, so the dose must be adjusted according to the renal function. Also, it is eliminated by hemodialysis, so it must be taken after the hemodialysis session. When CKD progresses, the classic therapeutic approaches present some peculiarities that are discussed below:
Erythropoietin analogsAs GFR drops, erythropoietin production decreases and anemia worsens. In these cases, although with little evidence, it is recommended the use of erythropoietin analogues in association with hydroxyurea.12,29 These patients often require higher doses than the used in other patients. The values of Hb should not exceed 10 g/dl due to the risk of promoting vaso-occlusive crises and other thrombotic phenomena caused by increased blood viscosity, however this level of Hb are rarely achieved, and most patients require periodic transfusions when they progress to end-stage kidney disease.12
DialysisA study that included 397 patients with sickle cell nephropathy on dialysis revealed that the mean age to need dialysis treatment was much lower in patients with SCD (40 vs 60 years), and that SCD was an independent risk factor for death, greater than diabetes, but that it cease to be so after kidney transplantation.38
Hemodialysis has the advantage of offering a route for transfusions, however when choosing the dialysis technique it should be considered, that SCD patients often have poor peripheral access. Both SCD and hemodialysis are risk factors for PH, so a prior evaluation is recommended. In many cases, peritoneal dialysis is often the technique of choice because it allows better tolerance of ultrafiltration.
Kidney transplantIn patients with SCD, median survival at 6 years after transplantation is 70%,39 and should be considered in patients with ESRD.29 Treatment with regular red cell replacement should be considered once the patient has been placed on the waiting list (or a few months before living donor transplantation) and be maintained while the graft is functioning. The most frequent serious complication of transplantation is sepsis, whose risk is increased by the hipoasplenia or functional asplenia, so some authors postulate avoid using antithymocyte globulin for treatment of rejection of the graft.12
Diagnostic considerationsThere is no consensus on the lab measurements that should be obtain for screening of sickle cell nephropathy or on their periodicity. However, it is recommended to evaluate renal function at least annually since the age of 10 years,8,29 although it could be useful to do it earlier.29 it would be advisable to examine the first urine in the morning using a urinary strip and determine, both in blood and urine, sodium, potassium, creatinine, osmolality, albumin and total proteins, including the calculation of the indices urinary protein/creatinine and albumin/creatinine, as well as cystatin C and bicarbonate in blood (venous). Albuminuria must be confirmed in the first morning urine or in two consecutive samples at any time.29
The calculation of GFR from creatinine using the CKD-EPI equation in adults or using the Schwartz formula in children may overestimate the GFR due to hyperfiltration and increased creatinine secretion in the proximal tubule.40 Therefore it may be more appropriate to consider the changes in serum creatinine or estimated GFR rather that the absolute values.8 Serum cystatin C, which is freely filtered in the glomerulus and it is not secreted by the renal tubules, has been shown to be a more accurate marker of renal function than creatinine and could detect a GFR defect earlier, but its routine use has not been validated. Furthermore, people with SCD tend to have a low lean mass, and cystatin C does not seem to be associated with muscle mass as it is the creatinine.41
The usefulness of β2-microglobulin as a marker of proximal tubular dysfunction in SCD has not been well studied; in a retrospective longitudinal study, the levels of β2-microglobulin did not differ significantly in 120 children with and without albuminuria.42
One strategic approach is to refer patients to Nephrology if the protein-creatinine ratio >50 mg/mmol (442 mg/g), evidence of persistent microscopic hematuria, an annual drop in estimated GFR > 10%, or an estimated GFR <60 ml/min/1.73 m2.12 The nephrology specialist must be familiar with the manifestations of SCD, and in it may be recommended that a single specialist collect all cases in each center. Other causes of proteinuria or hematuria must be evaluated. The usefulness of renal biopsy has not been evaluated, and the decision must be individualized, although it should be made in cases of nephrotic syndrome.12
Genetic modifiers of sickle cell kidney diseaseAs in other manifestations of SCD, genotype is an important modifier of the prevalence and severity of sickle cell nephropathy. Thus, sickle cell nephropathy is more severe in the forms that associate a greater amount of Hb S and hemolysis (Hb S/S and Hb S/β0-thalassemia), while it is milder in the heterozygous compound Hb S/C forms. and Hb S/β+-thalassemia).13,43 The coexistence of deletions affecting the α-globin genes (α-thalassemic trait) is associated with lower Hb S levels in sickle cell trait, and protects against hyposthenuria44; in sickle cell anemia decreases the prevalence of macroalbuminuria.45
As previously mentioned, Hb F interferes with the polymerization of Hb S. In the absence of hydroxyurea treatment, Hb F levels varies between 5 and 20% depending on the haplotypes linked to Hb S and in relation to certain polymorphisms.14 Thus, some polymorphisms in the gene for myosin 9 (MYH9), apolipoprotein L1 (APOL1), bone morphogenetic protein receptor type 1B (BMPR1B) or in heme oxygenase-1 (HMOX1) have been associated with an increased risk of albuminuria, decrease in GFR or progression to CKD.14
Kidney abnormalities in sickle cell traitIndividuals with sickle cell trait carry a single allele affected by the hemoglobinopathy S mutation, while the other allele is normal. As observed by renal microangiography, they present distortion and destruction of the vasa rectus, although considerably less than patients with SCD.46 However, according to a meta-analysis published in 2018, hematuria is 2 times more frequent than in SCD.17 This fact could be due to the higher prevalence of sickle cell trait, and the high hematocrit of the carriers. In this same study, a higher risk of proteinuria and CKD was also observed in carrier individuals as compared to the general population. Therefore, it is reasonable to study kidney function in individuals with sickle cell trait if they have other risk factors of CKD, such as diabetes or hypertension.17 Finally, it is important to note that medullary renal carcinoma has been described almost exclusively in people with sickle cell trait, although it can also occur in individuals with SCD. Renal medullary carcinoma is a rare entity with a very poor prognosis that usually presents in early adulthood, in the form of gross hematuria, flank pain, and weight loss. In ultrasound it can be seen as a cyst, and this finding should be evaluated by contrast CT or MRI.47
New mechanisms involved in kidney damageThe hemolysis that occurs in patients with SCD causes massive release of Hb and heme group that are able to go through the glomerular filtration barrier and accumulate mainly in proximal tubular cells. In these cells, both Hb and the heme group promote various adverse effects, such as mitochondrial damage, inflammation, fibrosis, oxidative stress, and apoptosis.48 The heme group and Hb give rise to reactive oxygen species that generate lipid peroxidation, DNA damage, and inflammation.49 It has been described that the heme group is capable to induce directly an inflammatory response in mice with SCD through its binding to the Toll-type receptor 4, present in endothelial, mesangial, tubular cells, and in podocytes and the subsequent activation of the transcription factor NF-kB.50 Moreover, as commented, hemo acts as a potent vasoconstrictor agent because it binds nitric oxide and prevents systemic and renal vasodilatation.51
The Hb and heme group are transported by haptoglobin and hemopexin respectively to the liver where they are degraded by heme oxygenase-1. In SCD, there is an acquired deficit of haptoglobin and hemopexin due to chronic hemolysis.52 However, the heme group is also eliminated through the glomerular barrier attached to α-1-microglobulin (A1M). It has been recently described that in both humans and mice with SCD, there is a compensatory increase in A1M and the A1M-hemopexin ratio is augmented up to 10 times, in association with markers of hemolysis and tubular damage (KIM-1, Kidney Injury Molecule-1 and NGAL, neutrophil gelatinase-associated lipocalin).53 The administration of hemopexin and haptoglobin reduces kidney damage in experimental models of SCD.52,53
Traditionally tubular cells have been considered the only cellular targets for the adverse effects of Hb in the kidney, however data obtained by our group show that podocytes can capture Hb, which alters its function and viability. In podocytes, Hb increases the production of reactive oxygen species and induces apoptosis, both in vitro and in experimental models of intravascular hemolysis and as has been observed in biopsies of patients with other disorders associated with intravascular hemolysis (hemolytic syndrome atypical uremic and paroxysmal nocturnal hemoglobinuria).54 However, it is unknown whether this mechanism of damage is also present in patients with SCD. In adition, the harmful effects mediated by Hb on podocytes or tubular cells were reduced by activating Nrf2 (Nuclear erythroid 2-related factor 2), a transcription factor that increases the antioxidant and anti-inflammatory response.54,55 Various inducers of Nrf2 have shown to be beneficial in different models of kidney disease due to their ability to reduce oxidative stress and inflammation.56 Therefore, it would be interesting to analyze its effect in patients with SCD since it could offer a therapeutic alternative in sickle cell nephropathy.
Future outlookThe growing presence of SCD in our country requires greater awareness in healthcare professionals. Renal manifestations are among the most frequent and severe in sickle cell disease, and this is also extensive to the sickle cell trait. It is necessary to deepen in the knowledge of its pathophysiology in order to develop specific therapeutic options that are adapted to the heterogeneity of renal involvement and to the interrelations between the different processes that coexist. It is necessary to have biomarkers that allow patients to be stratified according to the risk of progressing to CKD and to carry out earlier and more effective therapeutic interventions. Although in recent years new treatments have been developed to alleviate the disease at a systemic level, it is necessary to analyze the effects on kidney disease.
FinancingThis work has been funded by the Carlos III Health Institute (ISCIII)/FEDER (PI17/00130, PI17/01495, PI20/00375, PI20/00487 and SAF2015-63696-R), (The Spanish Ministry of Science and Innovation/State Investigation Agency (10.13039/501100011033) (RTI2018-098788-B-100, DTS17/00203, DTS19/00093 and RYC-2017-22369) (Co-funded by European Regional Development Fund/European Social Fund “A way to make Europe”/“Investing in your future”), Spanish Society of Nephrology (SEN), Consejería de Salud y Familias-FEDER, Junta de Andalucía (PIGE-0052-2020), Center for Biomedical Research in Diabetes and Associated Metabolic Diseases Network (CIBERDEM) and Cardiovascular (CIBERCV).
Conflicts of interestThe authors declare that they have no conflict of interest.
Please cite this article as: Payán-Pernía S, Ruiz Llobet A, Remacha Sevilla ÁF, Egido J, Ballarín Castán JA, Moreno JA. Nefropatía falciforme. Manifestaciones clínicas y nuevos mecanismos implicados en el daño renal. Nefrologia. 2021;41:373–382.



