

The term monoclonal gammopathy of renal significance (MGRS) comprises a group of diseases pathogenetically characterised by proliferation of a B-cell or plasma cell clone that synthesises and secretes a monoclonal immunoglobulin or its components (light and/or heavy chains), that may deposit and cause glomerular, tubular, interstitial and/or vascular damage. The importance of differentiating the term MGRS from other monoclonal gammopathies lies in the fact that diagnostic and therapeutic procedures aimed at controlling monoclonal protein synthesis and secretion can be indicated, irrespective of the classic criteria based on malignant tumour expansion. Renal pathology associated with MGRS is highly heterogeneous, and therefore renal biopsy should be considered a key diagnostic tool. A precise diagnostic approach, however, must also identify the monoclonal protein in plasma and/or in urine, together with a complete haematological study in order to determine the nature and extension of cell clones. Recent advances in the understanding of these entities have resulted in significant improvements in clinical course and survival in several forms of MGRS, although more studies and clinical experience are needed in order to delineate more effective therapeutic strategies. In this review, we summarise the main clinical and pathological features of MGRS, highlighting the most appropriate diagnostic approach and current therapeutic options.
Bajo el término gammapatías monoclonales de significado renal (GMSR) se engloban un conjunto de enfermedades que se caracterizan patogénicamente por la proliferación de un clon de linfocitos B o células plasmáticas que sintetizan y segregan una inmunoglobulina monoclonal o uno de sus componentes (cadenas ligeras o pesadas), con capacidad para depositarse y producir daño a nivel glomerular, tubular, intersticial o vascular. La importancia de discriminar el término GMSR radica en poder indicar procedimientos diagnósticos y terapéuticos dirigidos al control de la síntesis y secreción de las proteínas monoclonales independientemente de los criterios clásicos vinculados con la expansión tumoral maligna. La patología renal asociada a las GMSR es muy heterogénea, lo que confiere a la biopsia renal una consideración de prueba diagnóstica clave. La correcta investigación diagnóstica de una GMSR debe incluir, además, la identificación en plasma u orina de la proteína monoclonal y un estudio hematológico completo que determine la naturaleza y extensión del clon celular. Los avances en el conocimiento de estas entidades han permitido mejorar el curso evolutivo y la supervivencia en varias formas de GMSR, aunque son necesarios más estudios y experiencia clínica para delinear protocolos terapéuticos más efectivos. En la presente revisión se resumen las principales características clínico-patológicas de las GMSR, se detalla la aproximación diagnóstica más adecuada, así como las opciones terapéuticas disponibles en el momento actual.
Several clinical entities are grouped under the name monoclonal gammopathies (MGs). They share a clonal proliferation of B-cells or plasma cells with the capacity to produce and secrete large amounts of a unique type of immunoglobulin or a constituent part of it (monoclonal component). The monoclonal component may be constituted of a heavy chain (normally γ chain and, less frequently, α, μ, δ, ¿ chains) along with a light chain (κ or λ), isolated light chains and, in exceptional circumstances, only heavy chains.1
The range of diseases, clinical manifestations and adverse health effects and persistence of entities is not only related to neoplastic cell proliferation, but also to the damage produced by the deposit of these monoclonal proteins in different organs, or through more complex pathogenic mechanisms, including autoimmunity, inflammation and fibrogenesis.2–4
In 2003, the International Myeloma Working Group2 revised the criteria for the diagnosis and classification of the clinical entities that are grouped under the term MG. In accordance with these criteria, there are four different entities:
- 1.
MG of undetermined significance (MGUS): monoclonal component <30g/l, with proliferation of plasma cells in bone marrow (<10%) and lack of clinical evidence of myeloma, lymphoma or amyloidosis.
- 2.
Asymptomatic or quiescent myeloma: monoclonal component ≥30g/l, with proliferation of bone marrow plasma cells ≥10%, but without evidence of organs or tissues involvement and, absence of the typical tetrad of hypercalcaemia, renal involvement, anaemia and bone lesions.
- 3.
Symptomatic myeloma which requires involvement of organs or tissues and which can also present as non-secretory (with no secretory component of monoclonal proteins). In 2014, the following additional criteria were incorporated: the presence of ≥60% plasma cells in bone marrow, a involved/uninvolved serum free light chain ratio ≥100, or the existence of more than one focal lesion using advanced imaging techniques (computed tomography [CT], magnetic resonance imaging [MRI] or positron emission tomography with 18F-fluorodeoxyglucose [PET-CT]).5
- 4.
Solitary bone plasmacytoma, extramedullary plasmacytoma and multiple solitary plasmacytomas.
Approximately 60% of all MGs are MGUS.6 In MGUS, a clone of B-cells or plasma cells, which are generally not neoplastic, produces and secretes small quantities of a monoclonal immunoglobulin or its components (light or heavy chains).7,8
This entity is a relatively common finding in the adult population (prevalence of 0.7% in the general population, increasing to 3% in those over the age of 50 and to 5% in those above the age of 70),8 with an annual standardised incidence of 4–15 cases per 100,000 according to different studies,9,10 but which may peak at 169 cases per 100,000 in individuals older than 80.10
It is estimated that in MGUSs there is neoplastic transformation (myeloma or lymphoma) in 1% annually.11–13 The factors which have proven to be determinant risk factors for neoplastic transformation are the following11–13: abnormal kappa (κ) to lambda (λ) free light chains ratio, different monoclonal immunoglobulin component (light or heavy chains) or of IgA type, and monoclonal protein concentration ≥15g/l. If these three factors are present, the risk of neoplastic progression reaches 58% in 20 years, while it is only 5% if none of these characteristics is present.13
In addition to the risk of neoplastic transformation, it has been demonstrated that these patients are also three to five times more likely to suffer from kidney diseases,14 and it has been observed in some studies that 23% of patients with light-chain MGUS have kidney disease.15
In the 1980s, there were already reports that the pathological scope of an MGUS was not only limited to neoplastic transformation, but that the synthesis and secretion of monoclonal (M) proteins could also cause other pathological processes with systemic effects which may develop by different pathogenic mechanisms.16–18 The kidney is one of the most commonly involved organs in the clinical course of an MGUS.
Renal involvement is very common in symptomatic myeloma, while the main pathogenic mechanism of myeloma-associated nephropathy is intratubular precipitation of monoclonal proteins produced by neoplastic cells (“cylinder nephropathy”).19–22 In this case, the secretion of large quantities of the M-protein are required to produce a massive precipitation, and the pathogenic key in this nephropathy is conditioned by the high burden and aggressiveness of the tumour.21,22
Descriptions of different pathological renal processes related to MGs have been increasingly reported. This led to the term MG of renal significance (MGRS)1,23–28 being adopted in order to distinguish and remove the uncertainty that exists on the benign progression of other MGs.
The importance of differentiating the term MGRS lies mainly in indicating targeted diagnostic and therapeutic procedures to control the synthesis and secretion of M-proteins — if it is confirmed that these are pathogenically linked to the nephropathy — irrespective of the classic haematological criteria which are more closely linked to the spread of the malignant tumour.3,4,22,26
In this review, the main clinical–pathological characteristics of MGRSs are described, the most appropriate diagnostic approach is detailed and the therapeutic advances and future perspectives are also discussed.
Kidney disease associated with monoclonal gammopathiesKidney disease associated with MGRSs is highly heterogeneous, which means that the renal biopsy is considered a key diagnostic test.1,22,25–28 However, the concomitant presence of kidney disease of another aetiology may make the correct histological interpretation difficult in some cases and be a confounding factor.25
For a correct investigation and interpretation of the findings, the optical microscopy must be accompanied of immunofluorescence with a panel of antibodies against light chains and immunoglobulin isotypes, electron microscopy (EM), must also be performed as well.4,22,25,29 In some cases, more sensitive but complex techniques should also be used, such as immunoelectron microscopy (IEM),30 or laser microdissection followed by proteomic mass spectrometry,31–34 to confirm the composition of the deposits and where they are located.
Immunofluorescence is crucial in the diagnosis to establish the pathogenic link to blood dyscrasia and, therefore, the use of anti- (κ and λ) light-chain antibodies should be compulsory practice in the histological study of any renal biopsy.25
In cases in which there is no usable kidney tissue in the frozen sample for immunofluorescence, it is possible to carry out “rescue” techniques of the sample in paraffin (Pronase treatment) with a high likelihood of success for the immunohistochemical detection of light chains.35
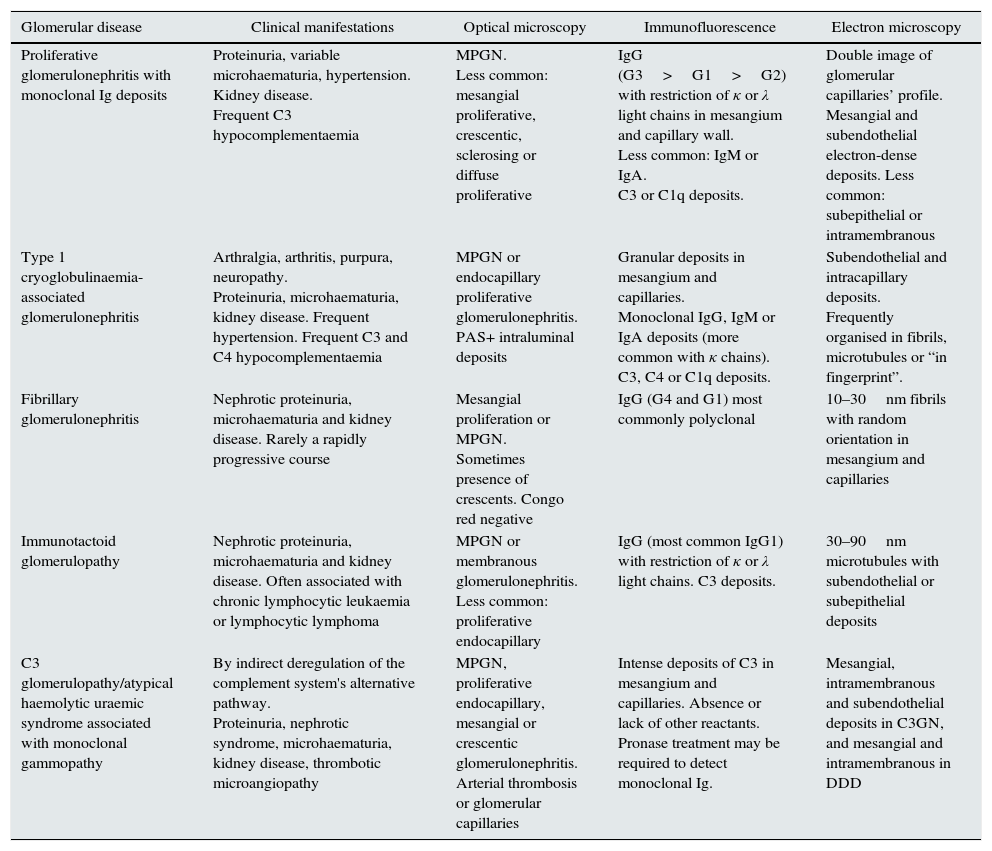
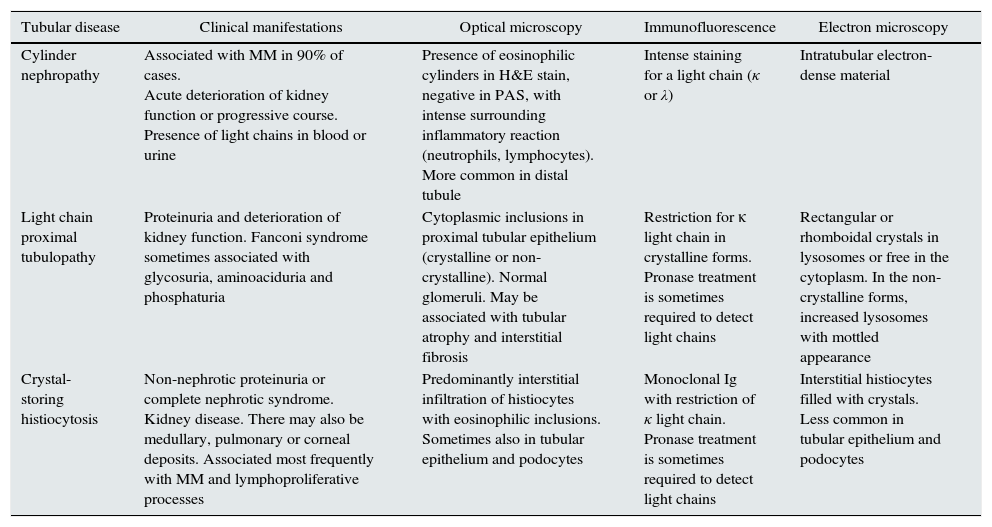
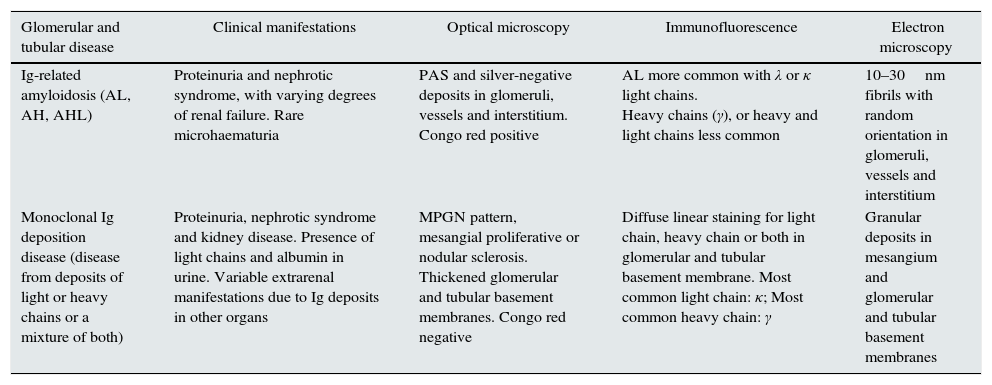
Different methods to classify the MGRSs have been proposed.1,4 One method considers the predominant location of the kidney damage (glomerular, tubular or mixed),1 although in practice it is not uncommon for more than one entity to overlap in the same biopsy.36–39 In Tables 1–3, the main clinical entities and their histological findings are summarised according to the extent of the damage.
Histological patterns of glomerular damage.
| Glomerular disease | Clinical manifestations | Optical microscopy | Immunofluorescence | Electron microscopy |
|---|---|---|---|---|
| Proliferative glomerulonephritis with monoclonal Ig deposits | Proteinuria, variable microhaematuria, hypertension. Kidney disease. Frequent C3 hypocomplementaemia | MPGN. Less common: mesangial proliferative, crescentic, sclerosing or diffuse proliferative | IgG (G3>G1>G2) with restriction of κ or λ light chains in mesangium and capillary wall. Less common: IgM or IgA. C3 or C1q deposits. | Double image of glomerular capillaries’ profile. Mesangial and subendothelial electron-dense deposits. Less common: subepithelial or intramembranous |
| Type 1 cryoglobulinaemia-associated glomerulonephritis | Arthralgia, arthritis, purpura, neuropathy. Proteinuria, microhaematuria, kidney disease. Frequent hypertension. Frequent C3 and C4 hypocomplementaemia | MPGN or endocapillary proliferative glomerulonephritis. PAS+ intraluminal deposits | Granular deposits in mesangium and capillaries. Monoclonal IgG, IgM or IgA deposits (more common with κ chains). C3, C4 or C1q deposits. | Subendothelial and intracapillary deposits. Frequently organised in fibrils, microtubules or “in fingerprint”. |
| Fibrillary glomerulonephritis | Nephrotic proteinuria, microhaematuria and kidney disease. Rarely a rapidly progressive course | Mesangial proliferation or MPGN. Sometimes presence of crescents. Congo red negative | IgG (G4 and G1) most commonly polyclonal | 10–30nm fibrils with random orientation in mesangium and capillaries |
| Immunotactoid glomerulopathy | Nephrotic proteinuria, microhaematuria and kidney disease. Often associated with chronic lymphocytic leukaemia or lymphocytic lymphoma | MPGN or membranous glomerulonephritis. Less common: proliferative endocapillary | IgG (most common IgG1) with restriction of κ or λ light chains. C3 deposits. | 30–90nm microtubules with subendothelial or subepithelial deposits |
| C3 glomerulopathy/atypical haemolytic uraemic syndrome associated with monoclonal gammopathy | By indirect deregulation of the complement system's alternative pathway. Proteinuria, nephrotic syndrome, microhaematuria, kidney disease, thrombotic microangiopathy | MPGN, proliferative endocapillary, mesangial or crescentic glomerulonephritis. Arterial thrombosis or glomerular capillaries | Intense deposits of C3 in mesangium and capillaries. Absence or lack of other reactants. Pronase treatment may be required to detect monoclonal Ig. | Mesangial, intramembranous and subendothelial deposits in C3GN, and mesangial and intramembranous in DDD |
Histological patterns of tubular damage.
| Tubular disease | Clinical manifestations | Optical microscopy | Immunofluorescence | Electron microscopy |
|---|---|---|---|---|
| Cylinder nephropathy | Associated with MM in 90% of cases. Acute deterioration of kidney function or progressive course. Presence of light chains in blood or urine | Presence of eosinophilic cylinders in H&E stain, negative in PAS, with intense surrounding inflammatory reaction (neutrophils, lymphocytes). More common in distal tubule | Intense staining for a light chain (κ or λ) | Intratubular electron-dense material |
| Light chain proximal tubulopathy | Proteinuria and deterioration of kidney function. Fanconi syndrome sometimes associated with glycosuria, aminoaciduria and phosphaturia | Cytoplasmic inclusions in proximal tubular epithelium (crystalline or non-crystalline). Normal glomeruli. May be associated with tubular atrophy and interstitial fibrosis | Restriction for κ light chain in crystalline forms. Pronase treatment is sometimes required to detect light chains | Rectangular or rhomboidal crystals in lysosomes or free in the cytoplasm. In the non-crystalline forms, increased lysosomes with mottled appearance |
| Crystal-storing histiocytosis | Non-nephrotic proteinuria or complete nephrotic syndrome. Kidney disease. There may also be medullary, pulmonary or corneal deposits. Associated most frequently with MM and lymphoproliferative processes | Predominantly interstitial infiltration of histiocytes with eosinophilic inclusions. Sometimes also in tubular epithelium and podocytes | Monoclonal Ig with restriction of κ light chain. Pronase treatment is sometimes required to detect light chains | Interstitial histiocytes filled with crystals. Less common in tubular epithelium and podocytes |
Histological patterns of mixed glomerular and tubular damage.
| Glomerular and tubular disease | Clinical manifestations | Optical microscopy | Immunofluorescence | Electron microscopy |
|---|---|---|---|---|
| Ig-related amyloidosis (AL, AH, AHL) | Proteinuria and nephrotic syndrome, with varying degrees of renal failure. Rare microhaematuria | PAS and silver-negative deposits in glomeruli, vessels and interstitium. Congo red positive | AL more common with λ or κ light chains. Heavy chains (γ), or heavy and light chains less common | 10–30nm fibrils with random orientation in glomeruli, vessels and interstitium |
| Monoclonal Ig deposition disease (disease from deposits of light or heavy chains or a mixture of both) | Proteinuria, nephrotic syndrome and kidney disease. Presence of light chains and albumin in urine. Variable extrarenal manifestations due to Ig deposits in other organs | MPGN pattern, mesangial proliferative or nodular sclerosis. Thickened glomerular and tubular basement membranes. Congo red negative | Diffuse linear staining for light chain, heavy chain or both in glomerular and tubular basement membrane. Most common light chain: κ; Most common heavy chain: γ | Granular deposits in mesangium and glomerular and tubular basement membranes |
The most accepted classification of lesions associated with MGRS is based on the distinction of the structure of deposits or inclusions according to whether these show an “organised” or “non-organised” configuration4,25 (Table 4).
Classification scheme of MGRS-associated disease according to the presence of organised or non-organised deposits.
| Organised deposits | Fibrils | Ig-related amyloidosis (AL, AH, AHL) |
| Fibrillary glomerulonephritis | ||
| Microtubules | Immunotactoid glomerulonephritis | |
| Type 1 cryoglobulinaemic glomerulonephritis | ||
| Crystals or inclusions | Proximal tubulopathy (with or without Fanconi syndrome) | |
| Crystal-storing histiocytosis | ||
| Non-organised deposits | Monoclonal Ig deposition disease (light or heavy chains or a mixture of both) | |
| Proliferative glomerulonephritis with monoclonal Ig deposits | ||
| Monoclonal Ig-associated C3 glomerulonephritis | ||
AH: heavy-chain amyloidosis; AHL: heavy- and light-chain amyloidosis; AL: light-chain amyloidosis; Ig: immunoglobulin.
Source: A modified version of the original scheme by Bridoux et al.4
“Organised” deposits are subdivided into: fibrils, microtubules and crystals or inclusions.
The pathological processes associated with fibrils are: immunoglobulin-related amyloidosis (of light chains, heavy chains or mixtures of light and heavy chains)4,25,40–43 and fibrillary glomerulonephritis.4,33,44,45
When the microstructure of the deposits adopts a microtubular structure, two entities can be distinguished4,25: immunotactoid glomerulonephritis, also known as glomerulonephritis with organised microtubules and monoclonal immunoglobulin deposits,33,44,46 and type 1 cryoglobulinaemia-associated glomerulonephritis.33,47–51
The crystal and inclusion deposits4,25 produce a predominantly tubular and interstitial disease, and two subtypes can be distinguished: proximal tubulopathy (with or without Fanconi syndrome)52–54 and histiocytosis with crystals stores,51,55–59 in which the crystal deposits are not found in the tubular epithelial cells, but instead inside the histiocytes.1 Cases of acute interstitial nephritis unrelated to the crystal deposits or intratubular precipitation of cylinders have also been reported, but with light chain deposits shown in the tubular basement membranes.60
Disease entities with “non-organised” deposits include: Randall-type monoclonal immunoglobulin deposit disease (disease caused by deposits of light chains, heavy chains or a combination of both)25,61–63; proliferative glomerulonephritis with monoclonal immunoglobulin deposits17,25,64–68 and monoclonal gammapathy-associated C3 glomerulopathy (C3G).69–74 In the case of proliferative glomerulonephritis with monoclonal immunoglobulin deposits, involvement is limited to the glomerulus, while in monoclonal immunoglobulin deposit disease there is extraglomerular and, frequently, extrarenal involvement.4
In addition to these processes, there are reports suggesting a possible pathogenic involvement of MG in other glomerulopathies, such as membranous GN,75,76 focal segmental glomerulosclerosis,77 pauci-immune extracapillary glomerulonephritis,78 C4 proliferative glomerulonephritis79 and in thrombotic microangiopathies.79–83
Clinical presentationMGRSs may show a wide range of manifestations, depending on the underlying pathogenic mechanism and the primary site of involvement.1,3,4 In the majority of cases, the deposit of M-proteins is responsible for the kidney disease, while in other cases it is caused indirectly through a deregulation of the complement system's alternative pathway, resulting in C3G,1,3,4,71 or, more rarely, in atypical haemolytic uraemic syndrome.81 Thus, the monoclonal component would be able to interfere with the regulatory proteins of the complement system, acting as miniautoantibodies against factor H,73,74 or as a C3 nephritic factor, which stabilises the C3 convertase of the alternative pathway and maintains its hyperactivation.1,70,71
The structural and chemical characteristics of each specifc M-protein, and the inflammatory response of each individual, seem to be key to determine the type of kidney damage being produced.18,84 High-molecular-weight proteins, such as immunoglobulins (formed by heavy and light chains), do not cross the filtration barrier and are deposited in the glomerulus which triggers an inflammatory processes. In contrast, light chains are able to cross the filtration barrier and generate a variety of tubular damage.
Renal involvement is often the first manifestation of blood dyscrasia.26 According to the different series, the age of diagnosis tends to be over 50. The disease of monoclonal immunoglobulin deposits is more common in males and proliferative glomerulonephritis with monoclonal immunoglobulin deposits is more common in women.63,66,85
Within the clinical manifestations of MGRSs, it is common to find different degrees of proteinuria, which may reach the nephrotic range, together with microhaematuria and hypertension in certain cases. Renal failure is detected in a high percentage of patients at the time of diagnosis, which may progress to end-stage kidney disease.4,63,66,86 This is of particular importance given that the recurrence of some of these diseases in transplanted kidneys is very common.87–92 This fact illustrate the need for a correct diagnosis, even when it is unlikely to preserve the function of the native kidneys.
Extrarenal manifestations may also be presented, especially in cases of amyloidosis, monoclonal immunoglobulin deposition disease and type 1 cryoglobulinaemia, with involvement predominantly of the heart, liver, skin and joints.1,4,93 The possibility of the disease spreading to other organs and tissues in patients diagnosed with amyloidosis should be investigated,43 due to the common nature of cardiac involvement, which tends to be the main determinant of mortality.43
Cases of osteomalacia secondary to Fanconi syndrome have also been reported.52
Other systemic manifestations of an MG may be related to endothelial damage and systemic thrombotic microangiopathy associated with the secretion of vascular endothelial growth factor (VEGF), as occurs in POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, MG and skin lesions)94 and scleromyxoedema.95
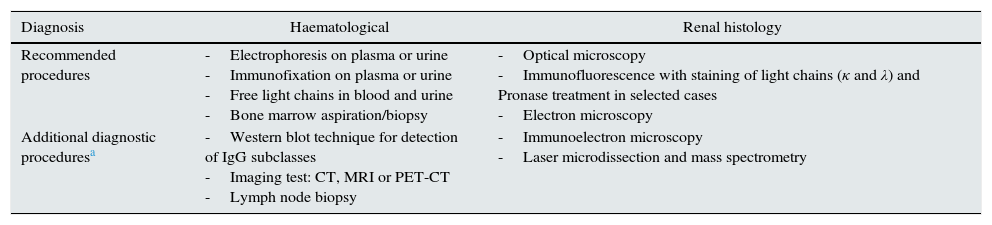
Diagnosis of monoclonal gammopathies of renal significanceThe appropriate diagnostic procedure of an MGRS should include, in addition to a renal biopsy, the demonstration and identification of MG in plasma or urine, a haematological study which determines the nature and extent of the cell clone causing the MG and, in some processes (e.g. amyloidosis), extension of the study to determine the spreading of the disease to other organs.4,25,26 The main techniques for the haematological and histological diagnosis are summarised in Table 5.
Techniques for the haematological and renal histology diagnosis of MGRSs.
| Diagnosis | Haematological | Renal histology |
|---|---|---|
| Recommended procedures | -Electrophoresis on plasma or urine -Immunofixation on plasma or urine -Free light chains in blood and urine -Bone marrow aspiration/biopsy | -Optical microscopy -Immunofluorescence with staining of light chains (κ and λ) and Pronase treatment in selected cases -Electron microscopy |
| Additional diagnostic proceduresa | -Western blot technique for detection of IgG subclasses -Imaging test: CT, MRI or PET-CT -Lymph node biopsy | -Immunoelectron microscopy -Laser microdissection and mass spectrometry |
CT: computed tomography; IgG: immunoglobulin G; MRI: magnetic resonance imaging; PET-CT: positron emission tomography with 18F-fluorodeoxyglucose.
The diagnosis of a suspected MGRS is almost always established by associating renal involvement (deterioration of function, proteinuria, Fanconi syndrome or other metabolic abnormalities associated with tubulointerstitial dysfunction) together with the presence of a monoclonal peak in the electrophoretic spectrum.4,96,97
In most cases, the diagnosis of MG is performed using conventional electrophoresis on plasma or urine.4,96 The presence of monoclonal immunoglobulins is usually identified by a tall narrow peak in the beta or gamma region, unlike polyclonal increase, which tends to create a wide band in the gamma region.96
However, in some cases the concentration of monoclonal protein in plasma or urine is so small that electrophoresis is unable to detect it. In fact, some cases of renal involvement in MG are diagnosed mainly by the findings in the renal biopsy (immunohistochemistry), without being suspected when the biopsy was indicated.
In addition to conventional electrophoresis, plasma and urine immunofixation must be performed in all cases to identify the type of M-protein, as this is more sensitive than electrophoresis in detecting the M-protein.4,96,98
Another diagnostic method is the determination of free light chains (FLCs) in blood and urine.96,98–102 The concentration of these proteins can be measured by a nephelometric immunoassay using polyclonal antibodies targeting light chain epitopes, which are exposed when the chain is in its free form but hidden when the chain is bound, creating the structure of the Ig.99
These measurements of FLCs are very sensitive, but the drawback is that they are not able to demonstrate the monoclonality of FLCs. This possibility is suggested indirectly by the ratio between the concentrations of the κ and λ chains.98,100
An abnormal ratio of κ and λ concentrations should be interpreted more carefully in patients with deterioration of kidney function.4,102 There is a strong correlation between the FLC concentration and kidney function; as the glomerular filtration rate decreases, the polyclonal FLC concentrations, of both κ and λ increase. Furthermore, in patients with normal kidney function, the greater physiological production of polyclonal κ FLC is masked by the more rapid clearance of monomeric forms of κ FLC, compared to the larger dimeric λ forms. This is how a change is produced in the ratio of κ and λ FLC concentrations in the case of renal failure.4,102 When kidney function is normal, the range of this ratio ranges from 0.26 to 1.65, while when kidney failure is present, the ratio accepted as normal ranges between 0.37 and 3.17, however a range for each stage of kidney failure has not been established.102
The difference in concentrations between FLCs is very useful not only for diagnosis, but also for follow-up and as a measure of the response to treatment. It is therefore recommended that it is monitored frequently.103
FLCs are also currently included in the treatment response criteria for AL amyloidosis.104 Therefore, a normalisation of the ratio of κ and λ concentrations, together with a negative result in the blood and urine immunofixation, is needed to be certain of a complete remission.
Using the FLCs measurement as an isolated test for detecting an MG is subject to debate105 and, although it may be useful to support the diagnosis of MG in myeloma, macroglobulinaemia and amyloidosis, the current recommendations state that electrophoresis (EP) and blood and urine immunofixation should be used for the diagnosis of MG.
More complex tests (western blot or electrotransfer) on blood and urine are required for the detection of some subclasses of immunoglobulins (IgG) or circulating monoclonal incomplete heavy chains. These tests are able to detect very small concentrations of monoclonal proteins with a high sensitivity,106 unfortunately these techniques are not routinely available.
In order to confirm the diagnosis of MGRS, it is not only necessary to prove the pathogenic involvement of MG in the kidney, but, in addition, myeloma must be ruled out and the cell clone producing the M-protein must be characterised because its relevance in the design of the therapeutic strategy. The support of a Haematology Department is needed for this.3,4,107
In cases of MGRS in which the renal biopsy shows IgG, IgA or LC, the bone marrow aspiration and biopsy use to be sufficient to demonstrate the clone using flow cytometry and immunohistochemical techniques.4 In addition, the spread of the bone tumour or the presence of solitary plasmacytoma should be ruled out with imaging techniques (CT, MRI or PET-CT).4,5 MRI is the technique with the greatest sensitivity in the detection of bone marrow infiltration, although it is laborious.5 PET-CT, on the other hand, is able to identify changes in the lesions throughout the follow up and avoids the use of intravenous contrast. However, it exposes patients to high levels of radiation and is more expensive.5
The possibility that a clone is from B-cells instead of from plasma cells should be suspected in patients with IgM MGRS. Then, the imaging study include areas suspected of hosting lymphadenopathies for biopsy.3
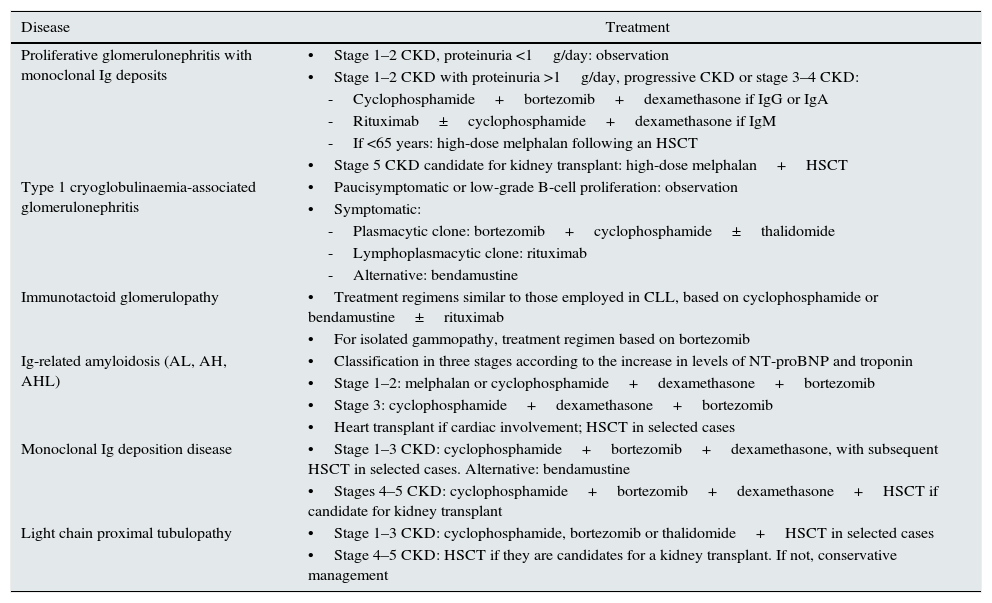
TreatmentThe treatment strategies for MGRSs are based on chemotherapy that must be adapted to the nature of the cell clone, both for lymphocytes and plasma cells, to the kidney function and to the presence or non-presence of extrarenal involvement.22,107
The rapid suppression of nephrotoxic monoclonal immunoglobulin has proven to be a treatment with satisfactory results on kidney function and patient survival in several forms of MGRS.107 However, as will be emphasised later, more studies and clinical experience are required to obtain therapeutic protocols based on solid evidence.
The fundamental objective of treatment, except for the case of light chain amyloidosis, must be aimed at preserving kidney function.26,91,107,108 This means that patients with irreversible kidney damage would not be candidates to receive chemotherapy treatment, unless there is a desire to obtain complete haematological remission to prevent recurrence in a kidney transplant.107
In 2013, the International Kidney and Monoclonal Gammopathy Working Group published a consensus document on the treatment regimens recommended for MGRSs, based on the clinical experience of the treatment of malignant blood dyscrasias.107Table 6 summarises the group's main recommendations. However, given the heterogeneity of MGRS-associated diseases, there is uncertainty regarding the optimal treatment for some more complex presentations with a clinical experience that is limited to isolated case reports or small series of cases.63,64,70,71,77,109 Thus, different regimens have been used for fibrillary glomerulonephritis, including cyclophosphamide, mycophenolate mofetil, cyclosporine, melphalan, lenalidomide and rituximab, with limited results.44,45,110 Furthermore, treatment regimens similar to those used for proliferative glomerulonephritis with monoclonal immunoglobulin deposits have been used for monoclonal gammopathy-associated C3 glomerulopathy.70,71,107 However, the information available on the long-term prognosis of these processes using the current therapies is insufficient.26
Therapeutic regimens proposed for MGRSs.
| Disease | Treatment |
|---|---|
| Proliferative glomerulonephritis with monoclonal Ig deposits | •Stage 1–2 CKD, proteinuria <1g/day: observation |
| •Stage 1–2 CKD with proteinuria >1g/day, progressive CKD or stage 3–4 CKD: | |
| -Cyclophosphamide+bortezomib+dexamethasone if IgG or IgA | |
| -Rituximab±cyclophosphamide+dexamethasone if IgM | |
| -If <65 years: high-dose melphalan following an HSCT | |
| •Stage 5 CKD candidate for kidney transplant: high-dose melphalan+HSCT | |
| Type 1 cryoglobulinaemia-associated glomerulonephritis | •Paucisymptomatic or low-grade B-cell proliferation: observation |
| •Symptomatic: | |
| -Plasmacytic clone: bortezomib+cyclophosphamide±thalidomide | |
| -Lymphoplasmacytic clone: rituximab | |
| -Alternative: bendamustine | |
| Immunotactoid glomerulopathy | •Treatment regimens similar to those employed in CLL, based on cyclophosphamide or bendamustine±rituximab |
| •For isolated gammopathy, treatment regimen based on bortezomib | |
| Ig-related amyloidosis (AL, AH, AHL) | •Classification in three stages according to the increase in levels of NT-proBNP and troponin |
| •Stage 1–2: melphalan or cyclophosphamide+dexamethasone+bortezomib | |
| •Stage 3: cyclophosphamide+dexamethasone+bortezomib | |
| •Heart transplant if cardiac involvement; HSCT in selected cases | |
| Monoclonal Ig deposition disease | •Stage 1–3 CKD: cyclophosphamide+bortezomib+dexamethasone, with subsequent HSCT in selected cases. Alternative: bendamustine |
| •Stages 4–5 CKD: cyclophosphamide+bortezomib+dexamethasone+HSCT if candidate for kidney transplant | |
| Light chain proximal tubulopathy | •Stage 1–3 CKD: cyclophosphamide, bortezomib or thalidomide+HSCT in selected cases |
| •Stage 4–5 CKD: HSCT if they are candidates for a kidney transplant. If not, conservative management |
AH: heavy-chain amyloidosis; AHL: heavy- and light-chain amyloidosis; AL: light-chain amyloidosis; CKD: chronic kidney disease; HSCT: haematopoietic stem cell transplantation; Ig: immunoglobulin; NT-proBNP: N-terminal pro-brain natriuretic peptide.
Source: Adapted from original by Fermand et al.107
Due to the growing importance of MGRSs in clinical nephrology, it is essential to be aware of the main therapeutic agents which are currently used, their tolerance and the most common side effects. Many of these treatments are administered in a combined form due to the synergistic effect on the B-cells and plasma cells.111 The main combinations include107,111,112: bortezomib, cyclophosphamide and dexamethasone; bendamustine and rituximab, and immunomodulatory agents, such as thalidomide and lenalidomide.
Bortezomib has a pivotal role within the therapeutic arsenal because of its safety profile and the possibility of being administered in full doses to patients with advanced kidney disease.112–114 The mechanism of action is based on the inhibition of proteasome activity, which produces apoptosis of the plasma cell and also inhibits the NF-κB pathway by reducing the release of proinflammatory cytokines and inducing anti-apoptotic pathways at the tubular level.115–117 Usually the side effects are not serious; it causes development or worsening of peripheral neuropathy, although this is less common when the route of administration is subcutaneous.118 Prophylaxis against herpes zoster is also recommended due to the risk of reactivation.111
Among the cytotoxic agents, both melphalan and cyclophosphamide have an effect on B-cells and plasma cells, however cyclophosphamide is used more frequently because it is less toxic.107,111 Another alternative is bendamustine, approved for the treatment of some lymphomas and suitable for patients with renal failure since it is predominantly metabolised in the liver.119–121
Melphalan is used in myeloablative doses as a conditioning therapy for autologous haematopoietic stem cell transplantation in selected cases with no significant extrarenal disease.107,111 This autologous transplant has been proven to improve survival of patients with multiple myeloma and light chain amyloidosis. However, in practice less than 20% of patients are suitable candidates for this treatment, which is associated with high morbidity and mortality.86,122–124
Monoclonal antibodies such as rituximab (targeting the CD20 antigen) are a suitable therapeutic option in the different forms of B-cell mediated MGRS, because of their good tolerance and limited number of side effects.107,111,112 The use of daratumumab (targeting CD38) for relapsed or refractory myeloma has been approved.125 However, experience on its use in cases of MGRS is currently lacking.
Within the family of immunomodulatory drugs, thalidomide would be more suitable than lenalidomide, due to the fact that the latter is excreted by the kidneys and can also cause kidney function deterioration in some cases.126–128 However, side effects have also been reported with thalidomide, such as the development of hyperkalaemia.129
ConclusionsMGRSs are associated with a wide range of kidney diseases resulting from the depositions of immunoglobulin or of its components in the kidneys, or through the deregulation of the complement system. Although the mortality of patients with MGRS is lower than that of myeloma or other related neoplastic forms, the likelihood of developing advanced chronic kidney disease is very high. For this reason, when evaluating patients with suspected MGRS it is necessary to conduct complete anatomopathological, haematological and biochemical studies. These studies allow to determine the type of entity and the extension of the disese. Advances in the understanding of these entities have made it possible to improve the clinical course and survival in several forms of MGRS. However, more studies and clinical experience are necessary to design more effective therapeutic protocols. It is a priority the close collaboration between nephrologists and haematologists to individualise the treatment to the clinical characteristics and comorbidity of patients, in an attempt to improve the overall prognosis of these diseases.
The incidence and prevalence of this group of diseases in the Spanish population is not currently known. There are few cases published.130 According to the clinical experience, these diseases are not frequent, but so far no epidemiological studies have been conducted which confirm these assessments. Furthermore, as they are rare diseases, but requiring a complex study, the diagnosis might end up being less appropriate or incomplete in patients attended to in many hospitals which do not have the necessary non-conventional diagnostic tools. Nevertheless the implementation of these tools in all hospitals does not seem to be an efficient measure. Therefore, the creation of centres of excellence and diagnostic–therapeutic reference for these types of diseases could be an appropriate investment to benefit from the health care, academic and economic value.
GLOSEN study proposalIn 2009, an international group for research into MGRSs was created. Nephrology, haematology and anatomical pathology departments from several countries participate in the group.4,26,107 The studies and publications by the members of this research group are currently recognised as the forefront of research into these diseases, with very significant diagnostic and therapeutic advances. The incorporation of Spanish groups into this international group could result in mutual benefits, such as training and transfer of experience to Spanish members, as well as the incorporation of Spanish centres and patients into international clinical trials.
All of these reasons could justify the creation of a national MGRS register which would help to research the clinical–pathological characteristics of these entities, and to identify evolutionary and response determinants to current treatments.
We believe that the ‘Grupo de Estudio de la Patología Glomerular de la Sociedad Española de Nefrología’ [Glomerular Disease Study Group of the Spanish Society of Nephrology] (GLOSEN) may be the ideal framework to lead a study of these characteristics, given its ample experience in conducting collaborative projects.131–133
As an initial project, it is proposed to collect retrospectively all diagnosed cases of MGRS with renal biopsy during recent years from the different centres taking part in the study. Although a work proposal will be sent to all members of the group soon, we are hereby launching a message to attract the interest of all nephrologists and pathologists interested in glomerular diseases, and requesting their collaboration in the study.
Conflicts of interestThe authors declare that they have no conflicts of interest related to the publication of this article.
- 1.
MGRSs are characterised by the proliferation of a clone of B-cells or plasma cells which synthesise and secrete a monoclonal immunoglobulin or one of its components (light or heavy chains), with the capacity to be deposited and be the cause glomerular, tubular, interstitial or vascular damage.
- 2.
Given the heterogeneity of MGRS-associated kidney disease, the renal biopsy is fundamental, and its proper histological analysis must include optical microscopy, immunofluorescence and electron microscopy.
- 3.
There are different ways of classifying MGRS-associated kidney disease, although the most accepted method is in accordance with the organisation of the deposits: “organised” (fibrils, microtubules and crystals) or “non-organised” (monoclonal immunoglobulin deposition disease, proliferative glomerulonephritis with monoclonal immunoglobulin deposits and monoclonal immunoglobulin-associated C3 glomerulonephritis).
- 4.
The diagnostic study should also include electrophoresis and plasma and urine immunofixation to identify the monoclonal protein and the determination of free light chains.
- 5.
In addition, myeloma should be ruled out and the cell clone producing the monoclonal protein should be characterised by a bone marrow aspiration and biopsy.
- 6.
Current treatment is based on clinical experience of the treatment of malignant blood dyscrasias. Close collaboration between nephrologists and haematologists to personalise the treatment to the clinical characteristics and comorbidity of patients is therefore a priority.
This study has been partly supported by the FEDER [Spanish Federation for Rare Diseases] funds ISCIII-RETIC REDinREN RD16/0009.
Please cite this article as: Caravaca-Fontán F, Gutiérrez E, Delgado Lillo R, Praga M. Gammapatías monoclonales de significado renal. Nefrologia. 2017;37:465–477.



