

Understanding the role of the complement system in the pathogenesis of atypical haemolytic uraemic syndrome and other thrombotic microangiopathies (TMA) has led to the use of anti-complement therapy with eculizumab in these diseases, in addition to its original use in patients with paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome. Scientific evidence shows that both primary and secondary TMAs with underlying complement activation are closely related. For this reasons, control over the complement system is a therapeutic target. There are 2 scenarios in which eculizumab is used in patients with TMA: primary or secondary TMA that is difficult to differentiate (including incomplete clinical presentations) and complement-mediated damage in various processes in which eculizumab proves to be efficacious. This review summarises the evidence on the role of the complement activation in the pathophysiology of secondary TMAs and the efficacy of anti-complement therapy in TMAs secondary to pregnancy, drugs, transplant, humoral rejection, systemic diseases and glomerulonephritis. Although experience is scarce, a good response to eculizumab has been reported in patients with severe secondary TMAs refractory to conventional treatment. Thus, the role of the anti-complement therapy as a new treatment option in these patients should be investigated.
El conocimiento del papel del complemento en la patogenia del síndrome hemolítico urémico atípico y otras microangiopatías trombóticas (MAT) ha fomentado el desarrollo de la terapia anticomplemento con eculizumab más allá de su indicación original en la hemoglobinuria paroxística nocturna y en el síndrome hemolítico urémico atípico. La evidencia científica demuestra un estrecho límite entre MAT primarias y secundarias con activación del complemento subyacente en ambas. Por ello, el control del complemento se convierte en una diana terapéutica. El uso de eculizumab en MAT secundarias contempla 2escenarios: diagnóstico diferencial difícil entre MAT primaria y secundaria (incluidos cuadros clínicos incompletos) o daño por complemento en procesos distintos, donde se demuestra la eficacia del tratamiento. Esta revisión es una síntesis de la evidencia científica sobre el papel de la activación del complemento en la fisiopatología de las MAT secundarias y la eficacia de la terapia anticomplemento en MAT asociadas a embarazo, fármacos, trasplante, rechazo humoral, enfermedades sistémicas y glomerulonefritis. La experiencia es aún limitada, pero la respuesta a eculizumab en pacientes con MAT secundarias graves y refractarias al tratamiento convencional abre una puerta a la investigación de la terapia anticomplemento como nueva opción terapéutica.
Research on the role of the complement system in the pathogenesis of atypical haemolytic uraemic syndrome (aHUS) and other thrombotic microangiopathies (TMAs) has stimulated the development of anti-complement therapy. Eculizumab, the first monoclonal antibody which blocks the complement component C5, is approved for the treatment of paroxysmal nocturnal haemoglobinuria and aHUS. Its efficacy in both native and transplanted kidney highlighted the importance of complement activation in aHUS, a prototype disease, caused by primary alteration of the complement system. However, diagnosis and academic classification by exclusion of different aetiologies and identification of genetic variants soon becomes more complex in light of evidence from situations in which different forms of TMAs converge, and the need to develop categories based on pathogenesis and treatment.
The first experiences which extended the original indication of eculizumab for paroxysmal nocturnal haemoglobinuria and aHUS are based on the scientific evidence of the narrow boundary between primary and secondary TMAs.1 Three concepts modify the classic view of TMA: primary or secondary complement activation, the overlapping in different clinical entities and endothelial dysfunction playing a key role in pathogenesis.
TMA is a complex process, resulting from the imbalance between immunity, clotting and complement, which is altered by precipitating factors (prevalent in secondary TMAs) in patients predisposed by multiple genetic determinants (dominant in aHUS) (Fig. 1).

Local activation of complement (aHUS, ischaemia/reperfusion) or systemic complement activation, is cause of endothelial lesion (humoral rejection, C3 glomerulonephritis [C3GN], recurrence of post-transplant nephropathy) is present in the primary or secondary form of TMA. Demonstration of the activation of the terminal effector complex (C5b-9) in secondary TMAs makes the complement system a therapeutic target and this is the basis of treatment with eculizumab2 in two scenarios: situations of a difficult differential diagnosis between primary and secondary TMA, and entities with activation of the terminal complement pathway with or without microvascular thrombosis:
- –
TMA difficult to diagnose:
- •
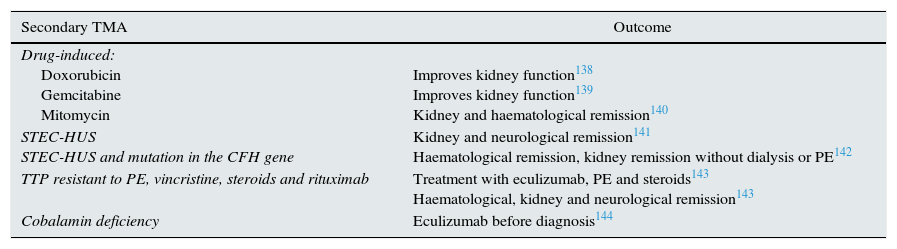
Triggered by infections (Table 1)
Table 1.Patients with complement-amplifying conditions treated with eculizumab.
Secondary TMA Outcome Drug-induced:
Doxorubicin
Gemcitabine
Mitomycin
Improves kidney function138
Improves kidney function139
Kidney and haematological remission140STEC-HUS
STEC-HUS and mutation in the CFH geneKidney and neurological remission141
Haematological remission, kidney remission without dialysis or PE142TTP resistant to PE, vincristine, steroids and rituximab Treatment with eculizumab, PE and steroids143
Haematological, kidney and neurological remission143Cobalamin deficiency Eculizumab before diagnosis144 PE: plasma exchange; STEC-HUS: Shiga toxin-associated E. coli-induced haemolytic uraemic syndrome; TTP: thrombotic thrombocytopaenic purpura.
- •
Associated with drugs (Table 1)
- •
Associated with glomerulonephritis
- •
In systemic diseases (vasculitis, lupus, scleroderma, etc.)
- •
In pregnancy and post-partum
- •
Malignant hypertension (MHTN)
- •
De novo post-solid organ transplantation (SOT-TMA)
- –
Microvascular damage with complement activation:
- •
TMA in haematopoietic stem cell transplantation (HSCT-TMA)
- •
Antibody-mediated rejection (AMR)
- •
Damage caused by ischaemia/reperfusion
- •
Refractory thrombotic thrombocytopaenic purpura
- •
Nephropathy caused by antiphospholipid antibodies.
Patients with TMA are identified quickly in clinical practice, but determining the cause is difficult. The time that elapses between suspicion and true demonstration of the disease may have a negative impact on the efficacy of the drug if the therapeutic decision is delayed. Genetic and molecular studies of the complement system have a long-term prognostic and strategic value, but not it is of limited help for early indication, which is decisive for preserving kidney function. TMA is a catastrophic and fatal process for the endothelium that, regardless of its cause, leads to kidney and systemic damage with a limited response to conventional therapy. Proof of this is that it TMA worsens the prognosis and survival of the patient in all situations in which it complicates the underlying disease: post-partum, kidney transplant and HSCT, MHTN, autoimmune diseases and glomerulopathies.
The objective of this review is to provide current scientific evidence on the treatment of secondary TMAs and the management of these patients from an emergent and promising therapeutic perspective.
Thrombotic microangiopathies in systemic diseasesThere are cases of TMAs associated with a great variety of systemic diseases, in which there is an overlap between entities and mutations of the complement system in up to 33% of patients with HUS associated with autoimmune diseases.3,4 The presence of diverse components of the complement system in kidney biopsies suggests its pathogenic role, and the response to eculizumab indicates that the deregulation of the non-genetic basis of the complement system may play an important role, and be a potential therapeutic target.
Antineutrophil cytoplasmic antibodies-associated vasculitisThe concept of pauci-immune vasculitis is changing due to the finding of electron-dense deposits in kidney biopsies in up to 54% of cases. In some patients with positive antineutrophil cytoplasmic antibodies-associated vasculitis, components of the complement system have been found in glomerular deposits (C3, C4, C1q, factor B, properdin and membrane attack complex) associated with a poorer kidney prognosis.5 TMA in kidney biopsies associated with vasculitis is not uncommon (13.6%), especially in severe cases and in cases with a worse outcome.6 It has recently been demonstrated that activation of the alternative complement pathway plays a key role in the pathogenesis of vasculitis.7 Experimental studies with a C5a receptor antagonist (CCX168) show a clear beneficial effect on the progression of the kidney condition.8 The use of drugs which block the complement system could therefore be a therapeutic alternative for these patients.
Systemic lupus erythematosusNumerous clinical observations reveal the importance of the complement system in lupus nephritis (LN), which histologically translates into the characteristic lesion known as “full house”, with immunoglobulin and complement deposits9 playing a double role in the pathogenesis of LN. The components of the classical pathway (C1q, C2, C4) play a protective role facilitating the clearance of SLE related immune complexes and apoptotic cells while final factors (C5 to C9) promote inflammation and tissue damage through the generation of anaphylatoxins (C5a) and the formation of the membrane attack complex (C5b-9).10 An experimental study revealed the role of factor H deficiency as an enhancer for the development of LN with a more aggressive clinical and histological presentation.11 The histological coexistence of LN and TMA results in a worse prognosis.12 Song et al. found TMA in 24.3% of kidney biopsies with LN in a retrospective study. These patients had more severe clinical expression and histological findings than the group that did not have TMA, resulting in a risk factor for deterioration kidney function.13 Experimental and clinical studies have shown that complement activation is essential in the pathogenesis of TMA and SLE. The use of complement blockers could therefore be a promising therapy.14 Treatment with eculizumab has proven to be safe and well-tolerated in phase I studies in patients with SLE; unfortunately, phase II or III studies have not performed.15,16 However, in the literature, five patients resistant to standard immunosuppressive therapy for LN (two associated with Catastrophic antiphospholipid syndrome (CAPS))responded positively to treatment with eculizumab (Table 2).10,17,18
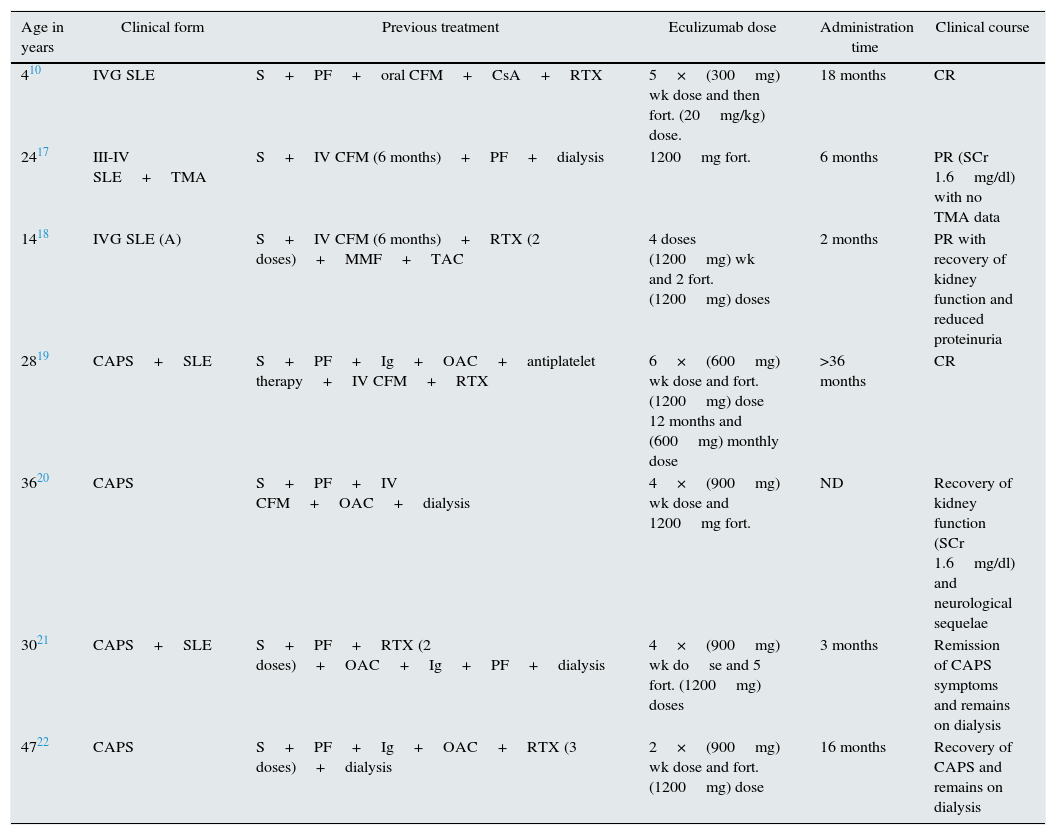
Patients treated with eculizumab in systemic diseases.
| Age in years | Clinical form | Previous treatment | Eculizumab dose | Administration time | Clinical course |
|---|---|---|---|---|---|
| 410 | IVG SLE | S+PF+oral CFM+CsA+RTX | 5×(300mg) wk dose and then fort. (20mg/kg) dose. | 18 months | CR |
| 2417 | III-IV SLE+TMA | S+IV CFM (6 months)+PF+dialysis | 1200mg fort. | 6 months | PR (SCr 1.6mg/dl) with no TMA data |
| 1418 | IVG SLE (A) | S+IV CFM (6 months)+RTX (2 doses)+MMF+TAC | 4 doses (1200mg) wk and 2 fort. (1200mg) doses | 2 months | PR with recovery of kidney function and reduced proteinuria |
| 2819 | CAPS+SLE | S+PF+Ig+OAC+antiplatelet therapy+IV CFM+RTX | 6×(600mg) wk dose and fort. (1200mg) dose 12 months and (600mg) monthly dose | >36 months | CR |
| 3620 | CAPS | S+PF+IV CFM+OAC+dialysis | 4×(900mg) wk dose and 1200mg fort. | ND | Recovery of kidney function (SCr 1.6mg/dl) and neurological sequelae |
| 3021 | CAPS+SLE | S+PF+RTX (2 doses)+OAC+Ig+PF+dialysis | 4×(900mg) wk dose and 5 fort. (1200mg) doses | 3 months | Remission of CAPS symptoms and remains on dialysis |
| 4722 | CAPS | S+PF+Ig+OAC+RTX (3 doses)+dialysis | 2×(900mg) wk dose and fort. (1200mg) dose | 16 months | Recovery of CAPS and remains on dialysis |
CAPS: catastrophic antiphospholipid syndrome; CFM: cyclophosphamide; CR: complete remission; fort.: fortnightly; Ig: immunoglobulin; IV: intravenous; MMF: mycophenolate mofetil; ND: no data: OAC: oral anticoagulant therapy; PF: plasmapheresis; PR: partial remission; S: steroids; SLE: systemic lupus erythematosus; RTX: rituximab; TAC: tacrolimus; wk: weekly.
Catastrophic antiphospholipid syndrome (CAPS) is a variant of antiphospholipid syndrome (<1%) characterised by systemic thrombosis and the development of multiple organ failure with elevated morbidity and mortality, and it is difficult to treat. Several authors have indicated that the uncontrolled activation of the complement system may initiate and amplify the characteristic phenomena of CAPS, such as endothelial activation, monocyte tissue factor expression and platelet aggregation, along with the histological findings of the TMA itself.19 Treatment ranges from anticoagulation to immunosuppressive therapy (steroids or cyclophosphamide), immunoglobulins and plasmapheresis. The use of therapies which block the complement system may be a therapeutic option, especially in patients who are refractory to standard treatment.20 In the literature, there are eight cases of CAPS who have been successfully treated with eculizumab, four of which are from transplant patients and four from non-transplant patients (Tables 2 and 3).19–27
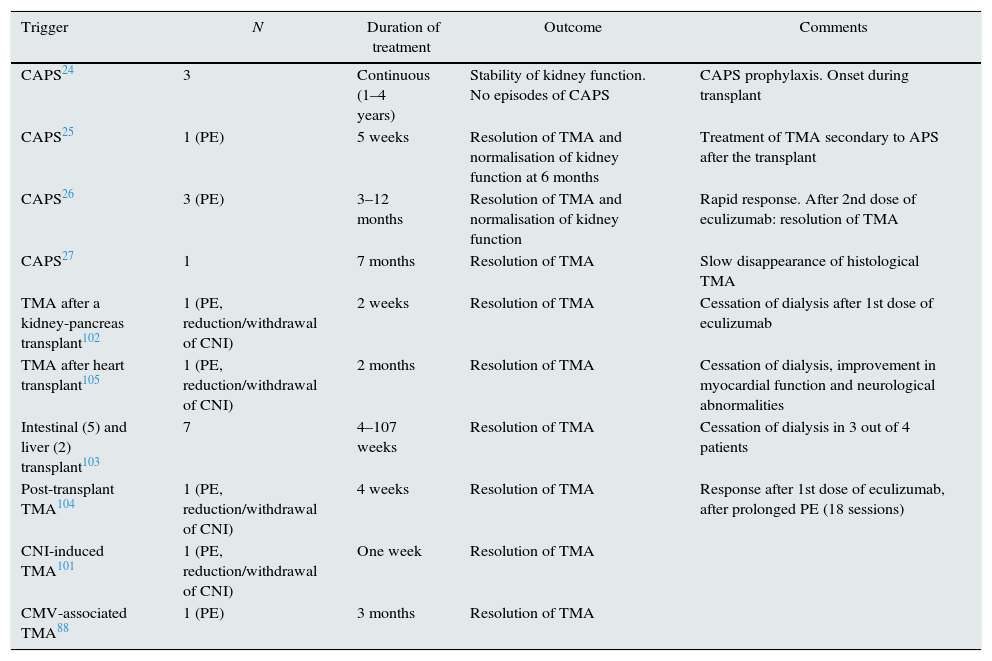
Use of eculizumab in TMA after solid organ transplantation.
| Trigger | N | Duration of treatment | Outcome | Comments |
|---|---|---|---|---|
| CAPS24 | 3 | Continuous (1–4 years) | Stability of kidney function. No episodes of CAPS | CAPS prophylaxis. Onset during transplant |
| CAPS25 | 1 (PE) | 5 weeks | Resolution of TMA and normalisation of kidney function at 6 months | Treatment of TMA secondary to APS after the transplant |
| CAPS26 | 3 (PE) | 3–12 months | Resolution of TMA and normalisation of kidney function | Rapid response. After 2nd dose of eculizumab: resolution of TMA |
| CAPS27 | 1 | 7 months | Resolution of TMA | Slow disappearance of histological TMA |
| TMA after a kidney-pancreas transplant102 | 1 (PE, reduction/withdrawal of CNI) | 2 weeks | Resolution of TMA | Cessation of dialysis after 1st dose of eculizumab |
| TMA after heart transplant105 | 1 (PE, reduction/withdrawal of CNI) | 2 months | Resolution of TMA | Cessation of dialysis, improvement in myocardial function and neurological abnormalities |
| Intestinal (5) and liver (2) transplant103 | 7 | 4–107 weeks | Resolution of TMA | Cessation of dialysis in 3 out of 4 patients |
| Post-transplant TMA104 | 1 (PE, reduction/withdrawal of CNI) | 4 weeks | Resolution of TMA | Response after 1st dose of eculizumab, after prolonged PE (18 sessions) |
| CNI-induced TMA101 | 1 (PE, reduction/withdrawal of CNI) | One week | Resolution of TMA | |
| CMV-associated TMA88 | 1 (PE) | 3 months | Resolution of TMA |
CAPS: catastrophic antiphospholipid syndrome; CMV: cytomegalovirus; CNI: calcineurin inhibitor; PE: plasma exchange; TMA: thrombotic microangiopathy.
Malignant hypertension (MHTN) is characterised by a marked increase in blood pressure and grade III or IV hypertensive retinopathy. Recently, it has been proposed to change this definition to severe hypertension with multiple organ failure.28 This entity may be accompanied by TMA with an estimated frequency of 5–20%.29,30 The hypotheses behind the presence of TMA in MHTN include: hyperstimulation of the renin–angiotensin–aldosterone system (elevated values of aldosterone as the main humoral mediator factor of TMA),31 genetic factors such as the TT genotype of angiotensinogen M235T and reduced values of ADAMTS13 in patients with MHTN and TMA.32,33 Furthermore, up to 15% of patients with TMA show clinical evidence of MHTN. It is essential to distinguish between these two entities as the treatment is different.34 Some clinical data may guide the differential diagnosis in favour of MHTN with TMA, such as a prior history of hypertension, excessively high values of blood pressure with deterioration of kidney function and mild or persistent thrombocytopaenia despite the control of blood pressure.35
Thrombotic microangiopathies in glomerulonephritisThe clinical, pathogenic and evolutionary characteristics C3 glomerulopathies (C3G) have been characterised in recent years. The definition is based on the presence of intense, isolated or clearly predominant deposits of C3 by immunofluorescence.36,37 Two types can be distinguished: C3GN and dense deposit disease; the latter is characterised by ribbon-shaped intensely osmiophilic deposits along the basement membrane.36 C3G pathogenesis consists of the abnormal activation of the alternative complement pathway by mutations in the genes coding for complement factors or regulatory proteins (factors H, I, CD46), or for autoantibodies against these regulatory factors.36–38
Various studies have shown that the spectrum of genetic mutations and autoantibodies associated with C3G is very similar to that of patients with aHUS.36–38 It is postulated that the deregulation of the complement system in C3G occurs in the fluid phase, resulting in the accumulation of the complement system's degradation products in the glomerular capillaries, while complement activation in aHUS mainly affects the cell surfaces (endothelium) and causes severe TMA.36,38–40
The reasons why some patients with a certain genetic mutation present with aHUS and others with C3G are only partially known.39,40 However, cases of aHUS/TMA and C3G as coincident processes41 in the same patient39,42–44 have been reported in the literature: TMA in patients previously diagnosed with C3G42 and others who develop aHUS/TMA after a kidney transplant,43,44 and whose end-stage kidney failure had been caused by C3G.
The treatment of C3G is widely debated. Although conventional immunosuppression had been considered ineffective based on some case reports and short series of patients, a recent study showed a favourable effect, especially for that based on steroids and mycophenolate mofetil (MMF).45 Preliminary data from this study indicate that the cases caused by autoantibodies could be especially sensitive to MMF, while those caused by genetic abnormalities would be, in general, resistant. Several patients have been treated with eculizumab, with varied results46–51; however a careful analysis of the cases indicates that eculizumab could be effective in patients with acute and aggressive disease, with no advanced chronic lesions in the kidney biopsy, and with increased serum levels of C5b-9.46–51 To date, we are not aware of any patient with C3G treated with eculizumab, who had developed TMA.
Immunoglobulin A nephropathyThe complement system plays a pivotal role in the pathogenesis of IgA nephropathy (IgAN), amplifying the kidney damage caused by the deposit of immune complexes composed of galactose-deficient IgA1 and its specific autoantibodies.52–54 Certain polymorphisms in the complement genes influence the predisposition to suffering from IgAN,52–54 and the deposits of C4d and C3 have a significant predictive value for this entity.55
The presences of TMA lesions in kidney biopsies of patients with IgAN have been indicated in some studies,56 but need to be corroborated. Several clinical cases have been reported showing TMA/aHUS and mutations in factor H associated with IgAN.57–59 Likewise, a beneficial effect of eculizumab in patients with aggressive IgAN without concomitant TMA/aHUS have been reported.60,61 It is clear that more studies are needed to determine the actual incidence of TMA in patients with IgAN, and the potential therapeutic indication of blockage of the complement system in this entity.
Other glomerulonephritisIntense deposits of diverse complement components, observed in most cases of glomerulonephritis, illustrate that in these processes complement activation plays a pivotal role in the glomerular damage. There are no systematic studies on the prevalence of genetic or functional abnormalities of the complement system in glomerulonephritis. Apart from C3G and IgAN, cases of TMA/aHUS have been reported in patients with focal segmental glomerulosclerosis, membranous nephropathy, acute post-infectious glomerulonephritis and immune complex-mediated membranoproliferative glomerulonephritis.62 There are no reports of cases treated with eculizumab in these TMAs associated with glomerulonephritis, although this drug has been used occasionally, with positive outcomes, in immune complex-mediated glomerulonephritis.63
Thrombotic microangiopathies in pregnancyPregnancy-associated TMA is a rare clinical entity with an estimated incidence of one case per every 25,000 pregnancies and with a high maternal and perinatal morbidity and mortality. During the last decade, two findings have contributed to improve the knowledge of this entity: the acquired or congenital deficiency of ADAMTS13, cause of thrombotic thrombocytopaenic purpura in the second and third trimesters of pregnancy, and the imbalance of regulatory factors of the complement system's alternative pathway as a genetic risk factor for the development of post-partum aHUS. Fakhouri et al. reported that 85% of patients with pregnancy-associated HUS presented abnormalities in the complement factors.64 Although the pathogenic mechanism need to be clarified, it is suggested that in a normal pregnancy with mutations in some the complement genes, there may be an uncontrolled activation counterbalanced by regulatory proteins located on the surface of the trophoblast such as CD55 or DAF (decay-accelerating factor), MCP and CD59,. However, post-partum TMA may be precipitated by numerous triggering factors (inflammatory phenomena, infections, haemorrhages) that could reactivate the complement system's alternative pathway after delivery without the presence of regulatory mechanisms on the placental surface.65 The involvement of the complement system in pregnancy-associated TMA has encouraged the use of eculizumab in seven patients, with or without complement mutations, during pregnancy or post-partum; different regimens were used, but an excellent clinical response was obtained in all cases (Table 4).66–71
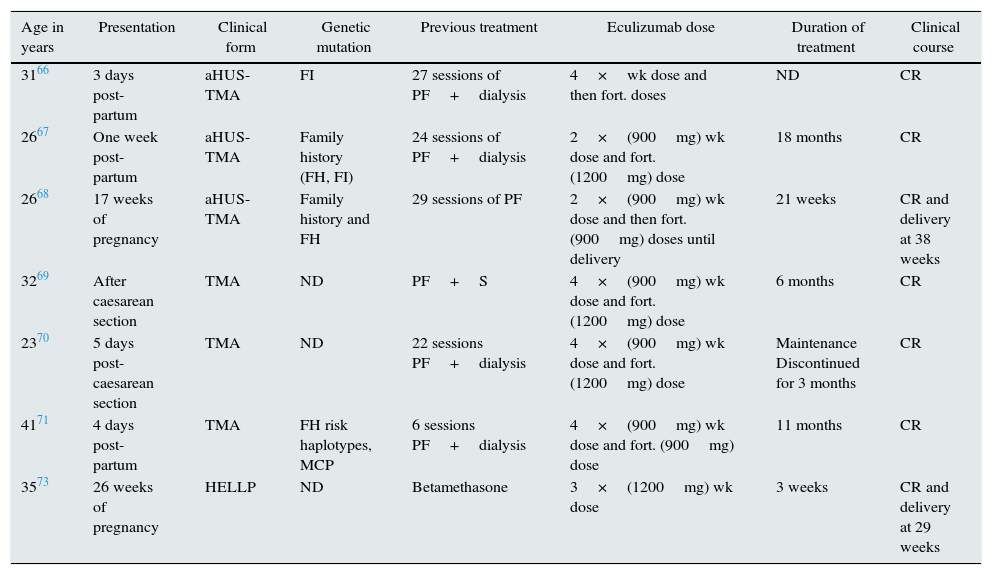
Patients treated with eculizumab in cases of pregnancy-associated TMA.
| Age in years | Presentation | Clinical form | Genetic mutation | Previous treatment | Eculizumab dose | Duration of treatment | Clinical course |
|---|---|---|---|---|---|---|---|
| 3166 | 3 days post-partum | aHUS-TMA | FI | 27 sessions of PF+dialysis | 4×wk dose and then fort. doses | ND | CR |
| 2667 | One week post-partum | aHUS-TMA | Family history (FH, FI) | 24 sessions of PF+dialysis | 2×(900mg) wk dose and fort. (1200mg) dose | 18 months | CR |
| 2668 | 17 weeks of pregnancy | aHUS-TMA | Family history and FH | 29 sessions of PF | 2×(900mg) wk dose and then fort. (900mg) doses until delivery | 21 weeks | CR and delivery at 38 weeks |
| 3269 | After caesarean section | TMA | ND | PF+S | 4×(900mg) wk dose and fort. (1200mg) dose | 6 months | CR |
| 2370 | 5 days post-caesarean section | TMA | ND | 22 sessions PF+dialysis | 4×(900mg) wk dose and fort. (1200mg) dose | Maintenance Discontinued for 3 months | CR |
| 4171 | 4 days post-partum | TMA | FH risk haplotypes, MCP | 6 sessions PF+dialysis | 4×(900mg) wk dose and fort. (900mg) dose | 11 months | CR |
| 3573 | 26 weeks of pregnancy | HELLP | ND | Betamethasone | 3×(1200mg) wk dose | 3 weeks | CR and delivery at 29 weeks |
aHUS: atypical haemolytic uraemic syndrome; CR: complete remission; FH: factor H; FI: factor I; fort.: fortnightly; ND: no data; PF: plasmapheresis; S: steroids; TMA: thrombotic microangiopathy; wk: weekly.
Also, complement activation has been recently associated with other entities related to pregnancy, such as pre-eclampsia and haemolytic anaemia syndrome, thrombocytopaenia and elevated liver enzymes (HELLP syndrome). Pre-eclampsia represents the maternal response to an excess of antiangiogenic factors generated by placental hypoperfusion which includes vasculopathic factors, such as Soluble Fms-Like Tyrosine kinase 1 (sFlt-1), a potent antagonist of vascular endothelial growth factor (VEGF) and endoglin (TGF-B inhibitor). The symptoms vary from different degrees of hypertension to HELLP syndrome or eclampsia.65,72 The complement system is key in inflammatory processes. Over activation of the complement system induces the deregulation of angiogenic factors which contribute to the inflammatory process. Based on this notion, Burwick et al. presented the case of a woman with severe HELLP syndrome at 26 weeks of pregnancy, treated with eculizumab, with a favourable clinical response. This made it possible to prolong the pregnancy by 17 days, reducing the likelihood of morbidity and mortality of the newborn.73
The complement system in solid organ transplantationsThrombotic microangiopathies occurring after kidney and other solid organ transplantsSOT-TMA is an uncommon complication observed in 0.8–15% of kidney transplants. It usually appears in the first three months (in 2/3 of patients) and results in graft loss in up to 1/3 of them.74–77
Clinical and microscopic characteristics of SOT-TMA are indistinguishable from recurrent aHUS.76 Personal and family history, abrupt onset and accentuated systemic and haematological involvement are more common in recurrent aHUS.3 Clinical presentation of SOT-TMA is variable; from isolated proteinuria and hypertension (30% of cases) to a full expression of TMA (microangiopathic haemolytic anaemia, thrombocytopaenia and deterioration of kidney function) with a greater risk of graft loss.74,76,78
As stated, aHUS is related to mutations in regulatory proteins of the complement system. In SOT-TMA there are multiple factors involved in endothelial damage and complement activation: donors with expanded criteria, events associated with brain death, ischaemia/reperfusion injury, viral infections, humoral rejection, antiphospholipid antibodies, anticardiolipin antibodies and anti-HCV and immunosuppressants, especially calcineurin inhibitors (CNIs) and mTOR inhibitors, intervene in SOT-TMA.26 Furthermore, genetic variants of complement proteins are observed in up to 30% of patients with SOT-TMA.74
In brain death and in ischaemia/reperfusion is associated with the release of C5a and C5b-9 and the capacity of CFH binding to the endothelium is reduced,26,78–83 this changes enhance the endothelial lesions observed in organs of donors who have had a cardiorespiratory arrest.84 Reduced tissue damage has been observed in experimental models when the complement system pathway is blocked.84,85 A clinical trial is being carried out to evaluate the effect of eculizumab on delayed graft function (NCT02145182).
Viral infections may trigger SOT-TMA and the recurrence of aHUS.86–89 Although the pathogenesis is unknown, it is related to the endothelial trophism of the virus that induces expression of adhesion molecules and release of the von Willebrand factor, which precipitates platelet adhesion and microvascular thrombosis.88 So far, there are seven cases reported of post-kidney transplant cytomegalovirus86–89; in all of them, the administration of intravenous ganciclovir and plasma exchanges managed to resolve the haemolysis. A recent publication has shown a second recurrence of cytomegalovirus viraemia-associated TMA resolved after treatment with valganciclovir, which facilitated the clearance of the viraemia, plus eculizumab, which can prevent cell destruction caused by the cytomegalovirus being mediated by the activation of the complement system.88
Immunosuppressant treatment is one of the main risk factors for SOT-TMA. This is observed in 4–15% of patients treated with cyclosporine and in 1–4% of those treated with tacrolimus.75,79,90 CNIs have a direct toxic effect on the endothelium which is mediated by the formation of microparticles stimulating the alternative C pathway and by tissue ischaemia (reduction of prostacyclin and nitric oxide vasodilators), formation of reactive O2 species and increased platelet aggregation.90–97 The mTOR inhibitors enhance post-transplant TMA by inhibiting VEGF synthesis by podocytes, enhancing endothelial damage and reducing endothelial regeneration capacity. The risk of SOT-TMA increases significantly when associated with both drugs.92,98–100
Management of SOT-TMA is not well established. Elimination of the causal agent (antiviral treatment, of the rejection, etc.) is initially recommended. In the case of drug-induced TMA, reducing or withdrawing the immunosuppressant may solve the TMA but increases the risk of acute rejection. In order to minimise this risk, changing the medication to belatacept could maintain the immunosuppression without increasing the risk of TMA.100 If TMA persists, or in severe cases, plasma exchange is recommended. Nevertheless, the risk of graft loss is 20–42%.74,77,78 Given the involvement of the complement system in post-transplant TMA and its poor prognosis, anti-complement therapy has been added to standard therapy in refractory cases in patients transplanted of different organs (Table 3).24,25,27,75,101–105 In a retrospective review, Dhakal reported a haematological recovery rate and an improvement in kidney function in 90% of 26 cases with HSCT-TMA and SOT-TMA treated with eculizumab. Although the dose, frequency and duration of treatment is variable (900 and 1200mg/dose and between 2 and 21 doses), the average response is observed after two doses (between 1 and 18).75
With a limited number of heterogeneous cases, this evidence supports the fact that eculizumab may be a therapeutic option for de novo post-SOT TMA, although more studies are needed to find out which group of patients would benefit, and what is the optimal dose and duration of treatment.
Antibody-mediated rejectionAntibody-mediated rejection (AMR) is a major problem in kidney transplanted patients with pre- and post-transplant human leucocyte antigen (HLA) antibodies. It may cause graft loss, rapid deterioration of kidney function and resistance to treatment. It is observed in 30–40% of sensitised patients despite immunosuppression, elimination of antibodies and splenectomy.
Eculizumab has been used in the prevention and treatment of AMR; its use is based on the critical pathogenic role of the complement system. Donor-specific antibodies activate the complement in the endothelium which triggers acute rejection and facilitates the inflammation process of chronic rejection. Ischaemia/reperfusion and immunosuppressants contribute to the amplification of the complement system, to the loss of endothelial resistance to the immune response and thrombosis.
The efficacy of eculizumab in AMR is not associated with histological TMA. Although this is found in 4% to 46% of patients, eculizumab not only increases the threshold for the development of TMA, but also interferes in the immune response. Specific blocking of C5 prevents the formation of C5a anaphylatoxin (potent inducer of inflammatory response) and the membrane attack complex C5b-9, which directly damages the endothelium.1
The strongest evidence supporting the powerful effect of complement control is the accommodation phenomena: acquired protection of the graft with positive C4d and normal histology. In the ABO-incompatible transplantation caused by anti-A/B antibodies, the expression of CD55 and CD59 regulatory proteins is increased. Even when used early after a transplant, eculizumab prevents acute rejection. There is no evidence of accommodation in treated patients.2
Eculizumab in the prevention of antibody-mediated rejectionThe most extensive experience has been published by Stegall et al.106 Twenty-six sensitised living donor transplant recipients received eculizumab to prevent AMR. When compared with 51 untreated former patients. The incidence of AMR was 7.7% in the treated group versus 40% in the untreated group. Two patients with high levels of donor-specific antibodies (DSAs) presented AMR during treatment.
Response to eculizumab varied in the different types of rejection. Bentall107 identified IgM DSA in four cases of the Stegall cohort: two of them with AMR and one with subclinical AMR. Burbach et al. published the case of two patients with poor progression in C4d-negative rejection.108
The development of IgM DSA, antibodies induced direct endothelial damage, antibody-dependent cellular cytotoxicity/complement-independent cellular cytotoxicity, activation of the alternative pathway or lectins and inflammatory lesions caused by proximal complement components could be mechanisms explaining the lack of response to eculizumab.
Eculizumab in the treatment of antibody-mediated rejectionThe literature includes a retrospective series including ten patients and ten independent cases.109 Orandi et al. treated 10 of 24 cases with severe antibody-mediated rejection (AMR) with eculizumab (with or without splenectomy).110 After a year, four of the five cases treated with eculizumab were lost to follow-up; none of the five treated with eculizumab plus splenectomy. Eight of another 10 independent cases responded to treatment and in two the graft was lost (failure at 47 days, BK nephropathy). A recently-published paediatric case underlines the importance of early treatment; treatment with two doses of eculizumab at an early stage improved kidney function and normalised histology.111
The pathogenesis of AMR is complex. Eculizumab does not have an impact on the levels of circulating DSAs or on the complement-independent cell response. It should therefore be combined with Thymoglobulin, rituximab, plasma exchange and IVIG. In Stegall et al.’s cohort, the duration of treatment varied (3–4 months). In this regard, the DSA level is important, and it is proposed to maintain treatment with MFI>9000.112
Eculizumab has proven to be effective in the prevention of AMR in sensitised patients, and in the treatment of refractory AMR. The impact on transplant glomerulopathy is difficult to assess due to the differences in severity of the AMR and the associated treatments. Early initiation is associated to greater efficacy, and it is necessary to gain more experience so the optimal duration of treatment in relation to the intensity of DSAs can be stabilised.
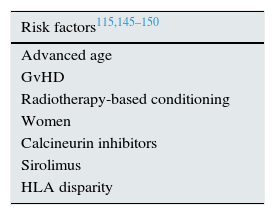
Haematopoietic stem cell transplantation-associated thrombotic microangiopathyHSCT-TMA is a complication with high mortality (up to 90% in patients with severe cases92) and with a risk of chronic nephropathy in patients with less severe cases. It has been estimated that TMA occurs in 10–35% of the haematopoietic stem cell transplantation (HSCTs), particularly after an allogeneic transplant.113–117 Triggering factors include CNIs for the prophylaxis of graft-versus-host disease, which directly cause endothelial lesion, activate the complement and alter the activity of ADAMTS13 (Table 5).97 Although acute graft-versus-host disease is an independent risk factor of HSCT-TMA, no causal relationship has been established.118–120
HSCT-TMA risk factors.
| Risk factors115,145–150 |
|---|
| Advanced age |
| GvHD |
| Radiotherapy-based conditioning |
| Women |
| Calcineurin inhibitors |
| Sirolimus |
| HLA disparity |
GvHD: graft-versus host disease; HLA: human leucocyte antigen.
HSCT-TMA may have a benign course without treatment or with a reduced dose of the CNI. However, some patients develop a systemic lesion with kidney injury, gastrointestinal damage, serositis, pulmonary hypertension and multiple organ failure.121–124 The most common endothelial lesion is the kidney lesion, which causes a decline in glomerular filtration rate, proteinuria and hypertension. Pulmonary involvement causes hypoxaemia or respiratory125,126 and gastrointestinal distress, vomiting, diarrhoea, abdominal pain and bleeding. This overlaps with the signs and symptoms of intestinal graft-versus-host disease, and requires a histological examination to reach a differential diagnosis.
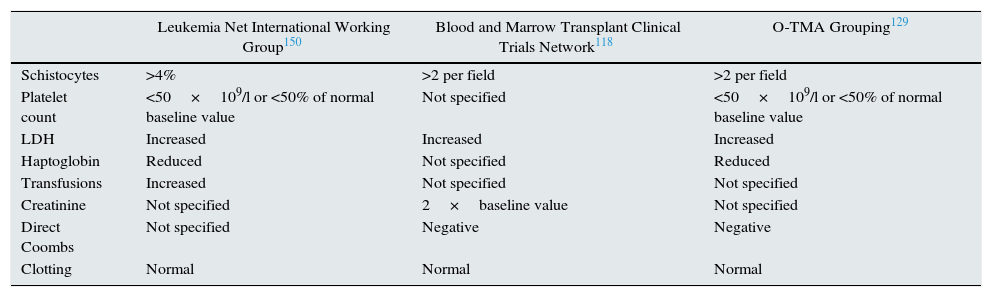
HSCT-TMA should be suspected if there is a sudden increase in serum lactate dehydrogenase (LDH) levels, hypertension and proteinuria. The increase in LDH is secondary to the ischaemic tissue damage related to thrombosis and endothelial lesion.127 The appearance of schistocytes is usually delayed. Table 6 lists the diagnostic criteria for HSCT-TMA.
Comparison of HSCT-TMA diagnostic criteria.
| Leukemia Net International Working Group150 | Blood and Marrow Transplant Clinical Trials Network118 | O-TMA Grouping129 | |
|---|---|---|---|
| Schistocytes | >4% | >2 per field | >2 per field |
| Platelet count | <50×109/l or <50% of normal baseline value | Not specified | <50×109/l or <50% of normal baseline value |
| LDH | Increased | Increased | Increased |
| Haptoglobin | Reduced | Not specified | Reduced |
| Transfusions | Increased | Not specified | Not specified |
| Creatinine | Not specified | 2×baseline value | Not specified |
| Direct Coombs | Not specified | Negative | Negative |
| Clotting | Normal | Normal | Normal |
LDH: lactate dehydrogenase; O-TMA: overall thrombotic microangiopathy.
The determination of the terminal complement activity by the quantification of plasma levels of the terminal effector complex (sC5b-9) would enable patients who may benefit from an anti-complement therapy to be identified. High plasma levels of sC5b-9 and proteinuria have been associated with a decline in survival rate (<20% after a year).128
Plasma levels of ADAMTS13, that normally are moderately reduced, can exclude thrombotic thrombocytopaenic purpura, which is extremely rare after an HSCT.
Lastly, in allogeneic transplant recipients, the genetic study of the complement system should be conducted in non-haematological samples, such as a buccal smear.
Initial treatment consists of removing potential triggering agents (CNI), controlling associated complications (infections, graft-versus-host disease) and an appropriate antihypertensive therapy. However, the response is often limited, especially in patients with severe HSCT-TMA. Current therapeutic options include plasma exchange, defibrotide, rituximab and eculizumab.
The use of plasma exchange is debated and is associated with a response rate of 36% (0–80%). This is probably because it is reserved for severe cases. The improvement of haematological parameters (platelets, haemoglobin, haptoglobin, LDH) may give a false impression of improvement of the underlying disorder. Complement regulatory proteins in infused plasma may produce remission in the short-term, but tissue damage and mortality are not modified.129,130 If plasma exchange is indicated, it should be started early and administered daily until the TMA is resolved.
The anticoagulant defibrotide, approved in Europe, has been used in patients with mild manifestations, and the administration of rituximab, alone or combined with other agents, has also been associated with a favourable response in selected cases.131
HSCT-TMA is a multifactorial disease in which the complement system is activated by the classical or alternative pathway, resulting in tissue damage caused by microvascular thrombosis.132 There is growing evidence of complement system imbalance involved in some cases of HSCT-TMA, and of complete remission with anti-complement therapy with eculizumab. It has been verified that patients with HSCT-TMA have complement activation, anti-factor H autoantibodies and C4d renal deposits,133–135 and may present pathogenic variants of aHUS complement genes. In a paediatric study of six children with acute kidney failure and HSCT-TMA, most presented with deletion of genes for complement factor H-related proteins (CFHR3 and CFHR1), three of them with anti-FH autoantibodies. Response to plasma exchange was poor, and high levels of sC5b-C9 and thrombosis were detected in the kidney biopsy. Of the six patients, four achieved therapeutic plasma levels of eculizumab and a clinical response.136
In a retrospective study in France from 2010 to 2013, 12 patients with severe HSCT-TMA received eculizumab, with a median follow-up of 14 months. The haematological and overall response was 50% and 33%, respectively.137
Both the doses of eculizumab and the period of time required to control HSCT-TMA are greater than the observed in aHUS,. It is recommended to assess the response after at least 4–6 weeks following the induction.
DiscussionThe involvement of the complement system in aHUS and TMA of distinct aetiologies uncover a new therapeutic perspective: control of the complement system to prevent endothelial and inflammatory damage. Experience shows that the range of TMAs is not made up of disjunctive categories, but that there is a continuum in the pathogenesis where two elements intervene and interact with variable intensity: genetic susceptibility and aetiological or predisposing factors. Scientific evidence shows overlap and common mechanisms between aHUS and other secondary TMAs in which complement-mediated endothelial damage occurs. This fact explains the positive response to eculizumab in patients with TMA of distinct aetiology, and makes it a promising therapy. The contribution of the complement system in the pathogenesis of secondary TMAs is well documented in the entities reviewed in this article. Systemic studies analysing the genetic variants and variants of predisposition to activation of the alternative and terminal pathways, the functional consequences of this activation on the disease mechanisms and the functional/clinical effect of blocking the C5 component by eculizumab, which represents a coadjuvant therapy to conventional treatment of the underlying disease in secondary TMAs, need to be carried out. Lack of response to the control of the aetiological factor and conventional treatment in secondary TMAs has supported the therapy with eculizumab in most of the published cases.
The fundamental limitations for evaluating the efficacy of eculizumab in secondary TMAs are: the clinical and pathogenic heterogeneity of generally severe and refractory cases; publication bias; concurrence of other therapies used in the underlying disease; dose variation; frequency and duration of treatments; and difficulty of monitoring using biological and pharmacokinetic markers. Optimal duration of treatment is an important issue, given the economic impact of eculizumab. This is also a limiting factor for early treatment which, as demonstrated in aHUS, is more cost-effective. The administration regimens correspond to the indication for aHUS and paroxysmal nocturnal haemoglobinuria in most cases, or they are modified empirically.
Although presently the overall number of patients treated is not very abundant, a significant number of cases are now treated successfully with eculizumab in post-pregnancy/partum TMA, post-SOT TMA, HSCT-TMA and in the prevention/treatment of AMR. Cases of TMA in systemic diseases associated with drugs, glomerulonephritis and MHTN form the most heterogeneous and difficult to evaluate group.
The involvement of complement activation in the pathogenic mechanisms of secondary TMAs is clear, but it is necessary to promote studies to standardise clinical experience, and to enable common strategies to be designed for different diseases and effective biological markers and clinical parameters to be identified, which make it possible to accurately determine the potential therapeutic indication in each case.
Key concepts- •
TMA is a complex process, derived from the imbalance between immunity, clotting and complement through a combination of aetiological factors (secondary TMAs) and genetic risk factors (dominant in aHUS).
- •
The classification of classic TMA does not explain the complexity of the disease mechanisms or reflect the therapeutic objectives. It is therefore necessary to reconsider a pathogenic classification of TMA.
- •
There is an overlap between both entities and common mechanisms of complement-mediated endothelial damage which explains the response to eculizumab in patients with TMA of distinct aetiology, making it a promising therapy.
- •
Although experience is limited, eculizumab has proven to be effective in post-SOT TMA, HSCT-TMA, prevention and treatment of humoral rejection, pregnancy-associated TMA and systemic diseases. As with aHUS, early treatment is associated with an improved therapeutic benefit.
- •
More studies are needed to accurately determine the indication, dosage and duration of treatment as a coadjuvant therapy to aetiological treatment in each case.
Elena Román, Javier de la Rubia, Amparo Sempere, Enrique Morales, Manuel Praga and Ana Ávila have carried out consulting and teaching activities for Alexion Pharmaceuticals. Elena Román, Enrique Morales and Manuel Praga have been members of the expert committees on aHUS. None of the above-mentioned activities has influenced the preparation of this manuscript.
Elena Román, Santiago Mendizábal, Isidro Jarque, Ana Ávila and José Luis Górriz are members of ‘Medicamentos de Alto Impacto Sanitario o Económico’ [Medicinal Products with a Major Health and Economic Impact] (MAISE) for eculizumab of the ‘Conselleria de Sanidad Universal y Salud Pública de la Generalitat Valenciana’ [Universal Healthcare and Public Health Council of the Regional Government of Valencia].
The authors would like to thank Alexion Pharmaceuticals for their logistical support in order to hold the meeting.
Please cite this article as: Román E, Mendizábal S, Jarque I, de la Rubia J, Sempere A, Morales E, et al. Microangiopatía trombótica secundaria y eculizumab: una opción terapéutica razonable. Nefrologia. 2017;37:478–491.



