



C3 glomerulonephritis is a rare, chronic disease characterized by C3c-dominant staining on renal biopsy and is caused by inherited or acquired alternative complement pathway dysregulation.
Case presentationHere, we reported a 36-year-old man presenting with nephritic syndrome and normal renal function. Secondary causes were excluded by detailed clinical history and laboratory tests. His renal biopsy was consistent with C3 glomerulonephritis with a membranoproliferative glomerulonephritis pattern. To identify the etiology, we carried out genetic and autoantibody screening tests. The results showed he was negative for autoantibodies, while the next-generation sequencing revealed common variants of complement factor H (c.1204T>C; p.Tyr402His), (c.184G>A; p.Val62Ile) and thrombomodulin (c.1418C>T; p.Ala473Val), which have previously been reported to increase susceptibility to complement-mediated diseases. He also carried complement factor H (c.2808G>T; p.Glu936Asp) and mannose-binding lectin (c.161G>A; p.Gly54Asp), putting the patient at an increased risk of infections, which was an important trigger for C3 glomerulonephritis. A novel variant of complement 2 (c.53A>G; p.His18Arg) that might contribute to the occurrence of C3 glomerulonephritis when combined with these susceptibility variants was further identified. The patient was treated with ramipril and regular fresh frozen plasma infusion. He had a good response to treatment with well-controlled proteinuria, stable renal function and an increasing serum C3 level.
ConclusionsThis case adds insight into the pathogenesis of C3 glomerulopathy by showing that a combination of susceptibility variants, genetic mutations and triggers might be responsible for the clinical and pathological phenotypes.
La glomerulonefritis C3 es una enfermedad rara y crónica caracterizada por tinción dominante en C3c en la biopsia renal y es causada por una desregulación de la ruta alternativa del complemento heredada o adquirida.
Presentación del casoInformamos sobre un varón de 36 años que presenta síndrome nefrítico y función renal normal. Las causas secundarias fueron excluidas por la historia clínica detallada y las pruebas de laboratorio. Su biopsia renal fue consistente con glomerulonefritis C3, con un patrón de glomerulonefritis membranoproliferativa. Para identificar la etiología realizamos pruebas de cribado genéticas y de autoanticuerpos. Los resultados mostraron que era negativo para autoanticuerpos, mientras que la secuenciación de próxima generación reveló variantes comunes del factor del complemento H (c.1204T>C; p.Tyr402His), (c.184G>A; p.Val62Ile) y trombomodulina (c.1418C>T; p.Ala473Val), que previamente se había informado que aumentaban la susceptibilidad a las enfermedades mediadas por el complemento. También tuvo factor del complemento H (c.2808G>T; p.Glu936Asp) y lectina de unión a manosa (c.161G>A; p.Gly54Asp), que aumentaba el riesgo de infección para el paciente, y que fue un desencadenante importante para glomerulonefritis C3. Una nueva variante del complemento 2 (c.53A>G; p.His18Arg) que podría contribuir a la aparición de glomerulonefritis C3 cuando se combina con estas variantes de susceptibilidad fue identificado. El paciente fue tratado con ramipril e infusión regular de plasma fresco congelado. Tuvo una buena reacción al tratamiento con proteinuria bien controlada, función renal estable y un aumento de suero del nivel C3.
ConclusionesEste caso agrega una idea de la patogénesis de la glomerulopatía C3 al mostrar que una combinación de variantes de susceptibilidad, mutaciones genéticas y desencadenantes podría ser responsable de los fenotipos clínicos y patológicos.
C3 glomerulopathy (C3G) is defined based on a common pathogenesis of excessive activation of the complement alternative pathway, manifesting as C3-dominant staining on renal biopsy after the exclusion of other diseases. On the basis of electron microscopic manifestations, C3G is subdivided into dense deposit disease (DDD) and C3 glomerulonephritis (C3GN). Several inherited and acquired causes have been recognized in C3G in previous studies.1,2 C3G clinically presents with different degrees of hematuria, proteinuria, hypertension and renal insufficiency. In addition, nearly 40–100% of DDD and 40–50% of C3GN patients showed a decreased serum C3 level with normal serum C4.3,4 The prognosis was variable, and a retrospective study indicated that age >16, DDD subtype and crescentic glomerulonephritis were independent predictors of end-stage renal disease (ESRD).4 Also, the latest study further found that estimated glomerular filtration rate (eGFR) and tubular atrophy/interstitial fibrosis were the strongest predictors for renal progression.5 Herein, we report a case of C3GN with combined genetic findings, indicating combined mutations with susceptibility variants might be useful for defining the risk of developing C3G.
Case presentationA 36-years-old Chinese man was admitted to Peking University First Hospital with a 5-years history of foamy urine. He started to have complaint of headache for 1 year, which was exacerbated 4 months prior to his admittance. Then, he was discovered to have malignant hypertension (204/151mmHg), and the funduscopic examination revealed flame hemorrhages and exudates. Urinalysis revealed proteinuria (3+) and dysmorphic red blood cells (5–8/high-power field). The 24-h proteinuria excretion was 3.76g, and the serum creatinine value was 1.04mg/dl (normal range: 0.50–1.50mg/dl). He was treated with ramipril (5mg/d) and nifedipine (30mg/d). His blood pressure was well controlled (110–120/70–80mmHg), his serum creatinine value was stable and the amount of proteinuria decreased to 2.94g/24h. Three months later, he maintained proteinuria (2.58g/24h) and was admitted to our hospital. He had no other systemic manifestations. He suffered from an episode of meningitis approximately 23 years ago. He denied any mental diseases and family history diseases. His parents were not consanguineous. He had never smoked, but sometimes he got alcoholic drunk on social occasions. He was not obese (body mass index 22.3kg/m2).
On admission, the physical examination revealed a blood pressure of 110/70mmHg, temperature 36.3°C, heart rate 80beats/min, and respiratory rate 20 breaths/min. The patient had mild bilateral symmetrical lower extremity edema. Other examinations were normal.
Laboratory data revealed proteinuria of 0.93–1.72g/24h, serum creatinine of 1.36mg/dl and serum albumin of 32.2g/l (normal range: 40–55g/l). His hemoglobin level was 134g/l (normal range: 130–175g/l), and the platelet count was 252×109cells/l (normal range: 125–350×109cells/l). Plasma C3 was 0.165g/l (normal range: 0.60–1.50g/l), and C4 was 0.177g/l (normal range: 0.12–0.36g/l). His serum immunoglobulin (Ig) G level was 8.05g/l (normal range: 7.23–16.85g/l), IgA was 1.01g/l (normal range: 0.69–3.82g/l), and IgM was 0.93g/l (normal range: 0.63–2.77g/l). C-reactive protein was <1.0mg/l (normal range: <8mg/l). Anti-streptolysin O, anti-neutrophil cytoplasmic antibodies, anti-glomerular basement membrane antibodies and anti-nuclear antibodies were all negative. HBsAg, anti-HCV, anti-HIV and TP-Ab were all negative. Chest X-ray and abdominal ultrasound were normal.
The patient underwent renal biopsy three days after hospitalization. Immunofluorescence examination revealed bright granular staining for C3c (+++), weak staining for IgA (+), IgM (+), and trace C1q along the glomerular capillary wall and in the mesangium (Fig. A1) but negative staining for IgG. Light microscopic examination exhibited that 5/32 glomeruli were globally sclerosed. Other glomeruli showed moderate to severe mesangial expansion with a lobular appearance, and segmental thickening of the glomerular basement membrane with double contours (Fig. A2). By electron microscopy, diffusely subendothelial and mesangial electron-dense deposits were identified, with extensive effacement of the foot processes of podocytes (Fig. A3).
. A2: Severe mesangial proliferation and thickening of the glomerular capillary wall with double contour formation, giving the glomeruli a lobular appearance (Periodic acid staining, 400×). A3: Mesangial and subendothelial electron-dense deposits under EM (8000×). Fig. B. Schematic presentation of the patient")
Renal biopsy findings of the patient. A1: Granular C3c deposition along the capillary wall and in the mesangium (indirect immunofluorescence staining on frozen tissue, 400×). A2: Severe mesangial proliferation and thickening of the glomerular capillary wall with double contour formation, giving the glomeruli a lobular appearance (Periodic acid staining, 400×). A3: Mesangial and subendothelial electron-dense deposits under EM (8000×). Fig. B. Schematic presentation of the patient's clinical course and treatment. Abbreviations: UTP: urine total protein excretion; SCr: serum creatinine; C3: complement 3.
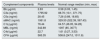
Then, we evaluated the complement activation status. The plasma levels of complement components were summarized in Table 1. He was negative for C3 nephritic factor (C3Nef), anti-CFH antibodies, anti-C3b antibodies, anti-CFB antibodies and monoclonal immunoglobulins. An integrated screening of 86 genes from the complement, coagulation and endothelial systems by using targeted genomic enrichment and massively parallel sequencing (Shanghai Biotechnology Corporation, China) revealed one homozygous common variant of CFH rs1061170 (NM_000186:chr1: 196659237; c.1204T>C; p.Tyr402His), two heterozygous common variants of CFH rs1065489 (NM_000186:chr1: 196709774; c.2808G>T;p.Glu936Asp) and rs800292 (NM_000186:chr1: 196642233; c.184G>A; p.Val62Ile), one homozygous common variant of thrombomodulin (THBD) rs1042579 (NM_000361:chr20: 23028724;c.1418C>T;p.Ala473Val), one heterozygous common variant of MBL2 rs1800450 (NM_000242: chr10:54531235;c.161G>A;p.Gly54Asp) and one novel heterozygous variant of complement 2 (C2) (NM_001145903: chr6:31901393;c.53A>G;p.His18Arg) after variant calling, quality control and hard filtering.6 All the variants were confirmed by Sanger sequencing.
Plasma levels of complement components in patient with C3 glomerulonephritis.
| Complement components | Plasma levels | Normal range median (min, max) |
|---|---|---|
| Bb (μg/ml) | 2.63 | 0.58 (0.03, 1.45) |
| C3a (ng/ml) | 1795.92 | 88.70 (10.1, 371.75) |
| C5a (ng/ml) | 28.45 | 7.26 (0.89, 18.65) |
| sMAC (ng/ml) | 1061.8 | 329.50 (322.39, 567.43) |
| C1q (μg/ml) | 76.44 | 64.57 (46.05, 94.28) |
| MBL (ng/ml) | 342.89 | 1097 (85, 3077) |
| C4d (μg/ml) | 22.7 | 1.61 (0.27, 3.53) |
| CFH (μg/ml) | 565.35 | 508.8 (247.0, 1011.8) |
Note: sMAC: soluble membrane attack complex; MBL: mannose-binding lectin;
CFH: complement factor H.
The patient was finally diagnosed as C3GN with a MPGN pattern. He did not have any complement-targeting autoantibodies or active changes in renal biopsy, so immunosuppressive agents were not initiated. He received 400–800ml fresh frozen plasma infusion monthly instead of 10–15ml/kg every 14 days as previous reported7 because of limited plasma supply. Ramipril was increased to 10mg/d, and nifedipine was stopped. His blood pressure was well-controlled (100–120/65–80mmHg), proteinuria decreased to less than 1g/d, serum creatinine was stable, and serum C3 increased to 0.4g/l. The plasma infusion was stopped after one and a half years because of plasma infusion risks and the patient's stable condition. His serum C3 was maintained at approximately 0.5g/l with slightly increasing serum creatinine (Fig. B).
DiscussionOur patient's renal biopsy exhibited C3c-dominant deposition on immunofluorescence and had no evidence of other diseases. Complement alternative pathway was over-activated with decreased plasma C3 and elevated plasma Bb, C3a, C5a and sMAC levels. Thus, a definitive diagnosis of C3G was made. The electron microscopy further excluded the subtype of DDD, giving this patient a diagnosis of C3GN with an MPGN pattern.8
The alternative pathway activation in C3GN was driven by inherited or acquired defects. Acquired causes included C3Nef, anti-CFH autoantibodies, anti-C3b autoantibodies or anti-CFB antibodies. Genetic factors included CFH, complement factor I (CFI), membrane cofactor protein (MCP), complement factor B (CFB), and C3 as well as CFH-related protein 5 (CFHR5) internal duplication, CFHR1-3 hybrid, CFHR1 duplication and CFHR2-5 hybrid.1,9–11 Up to date, reported variants associated with C3G in complement pathway from the four largest C3G cohort studies included plenty with no mutational hotspot, which was summarized in Table 2.1,5,12,13 C3GN has multiple clinical and pathologic phenotypes. The varied therapeutic strategies and prognoses are attributed to the different pathogeneses. Thus, it is important to identify the precise causes after the diagnosis of C3GN in order to guide individualized treatment. Herein, we conducted thorough inherited and acquired screening tests for this patient. As a result, he had no autoantibodies, but he was found to harbor multiple genetic changes, including common variants of CFH, THBD, and MBL2 and a novel variant of C2.
The complement gene variants identified from the four largest C3G cohort studies.
| Country | Case number | Genes screened | Genetic variants percentage | Variants identified | Histology | ExAC global frequency | Functional significance |
|---|---|---|---|---|---|---|---|
| Italy12 | DDD (n=21) | C3 | Mutations: | CFH p.Arg78Gly | C3GN | 0 | Likely pathogenic |
| C3GN (n=52) | CFB | C3: 7 (9.6%) | CFH p.Arg127Cys | C3GN | 0 | Likely pathogenic | |
| CFH | CFB: 2 (2.7%) | CFH p.Tyr1008X | C3GN | 0 | Likely pathogenic | ||
| CFI | CFH: 5 (6.8%) | CFH p.Arg1210Cys | DDD | 0.0002 | Likely pathogenic | ||
| MCP | CFI: 3 (4.1%) | CFH p.Thr956Met | C3GN | 0.0012 | Previously reported in aHUS2 | ||
| THBD | THBD: 2 (2.7%) | CFI c.1-4C>T | C3GN | 8.2E−06 | Likely pathogenic | ||
| CFI p.Gly57Asp | DDD | 8.2E−06 | Likely pathogenic | ||||
| CFI p.Arg317Trp | C3GN | 9.9E−05 | Previously reported in aHUS31 | ||||
| C3 p.Val619Met | C3GN | 0.0003 | Likely pathogenic | ||||
| C3 p.Arg1042Gln | C3GN | 0 | Likely pathogenic | ||||
| C3 p.Arg1303His | C3GN | 8.2E−06 | Likely pathogenic | ||||
| C3 p.Arg1320Gln | C3GN | 0 | Likely pathogenic | ||||
| C3 p.Cys1518Arg | C3GN | 0 | Likely pathogenic | ||||
| C3 p.Asp1625His | C3GN | 0 | Likely pathogenic | ||||
| C3 p.Lys633Arg | C3GN | 0.0004 | Previously reported in aHUS32 | ||||
| CFB p.Glu566Ala | C3GN/DDD | 0.011 | Previously reported in C3G2 | ||||
| THBD p.Asp486Tyr | C3GN | 0.0065 | Previously reported in aHUS33 | ||||
| THBD p.Pro495Ser | DDD | 0.0004 | Likely pathogenic | ||||
| France1 | DDD (n=29) | CFH | Mutations: | CFH p.Ala161Ser | DDD | 0 | Previously reported in P-HUS16 |
| C3GN (n=56) | CFI | CFH:12 (14.1%) | CFH p.Cys431Ser | DDD | 0 | Quantitative CFH deficiency | |
| MCP | CFI: 3 (3.5%) | CFH p.Arg232X | DDD | 0 | Quantitative CFH deficiency | ||
| MCP: 1 (1.2%) | CFH p.Val143Ile | DDD | 0 | Quantitative CFH deficiency | |||
| CFH p.Cys673Arg | DDD | 0 | Quantitative CFH deficiency | ||||
| Rare variants: | CFH p.Cys597Arg | GNC3 | 0 | Quantitative CFH deficiency | |||
| CFH: 2 (2.4%) | CFH c.230delT, Leu77X | GNC3 | 0 | Quantitative CFH deficiency | |||
| CFI: 3 (3.5%) | CFH c.363del21nt, del Gly122-Glu128 | GNC3 | 0 | Undetermined significance | |||
| MCP: 1 (1.2%) | CFH IVS 11+5 | GNC3 | 0 | Undetermined significance | |||
| CFH p.Asp130Asn | GNC3 | 0 | Likely pathogenic | ||||
| CFH p.Arg1210Val | GNC3 | 0 | Previously reported in aHUS34 | ||||
| CFH p.Arg53Cys | GNC3 | 0 | Previously reported in P-HUS16 | ||||
| CFI p.Ala240Gly; CFI p.Ala222Gly | GNC3 | 0 | Previously reported in HUS31 | ||||
| CFI p.Gly261Asp, CFI p.Gly243Arg | GNC3 | 0 | Previously reported in aHUS35 | ||||
| CFI p.Ile306Ser, Ile288Ser | GNC3 | 0 | Previously reported in aHUS35 | ||||
| MCP p.Val215Met,Val181Met | GNC3 | 0 | Undetermined significance | ||||
| MCP p.Ala304Val | GNC3 | 0 | Pathogenic, previously reported in aHUS36 | ||||
| The United States5 | DDD (n=9)C3GN (n=42) | C3CFBCFHCFIMCPCFHR5 | Genetic variants:C3: 2 (4.8%)CFH: 11 (21.6%)CFI: 0 (0.0%)CFB: 0 (0.0%)CFHR5: 1 (2.4%)MCP: 1 (2.4%) | Detailed information of variants was not provided | |||
| Spain13 | C3GN (n=60) | C3CFBCFHCFIMCP | Mutations(in 23 patients):C3: 1 (4.3%),CFH: 1 (4.3%) | Detailed information of variants was not provided | |||
Note: C3G: C3 glomerulopathy; DDD: dense deposit disease; C3GN: C3 glomerulonephritis; GNC3: glomerulonephritis with isolated C3 deposits; aHUS: atypical hemolytic uremic syndrome; C3: Complement C3; CFB: complement factor B; CFH: complement factor H; CFI: complement factor I; MCP: membrane cofactor protein; CFHR5: complement factor H-related protein 5; THBD: thrombomodulin; ExAC Global Frequency: variant frequency in all subjects of the ExAC database.
CFH is one of the key inhibitors of the alternative pathway. The CFH Val62Ile variant (rs800292) and Tyr402His variant (rs1061170), which locate within regions that bind C3b, heparin and C-reactive protein, have been shown to confer susceptibility to DDD patients.1,12 Meanwhile, the Tyr402His variant had significant association with an increased risk of developing age-related macular degeneration (AMD), another complement-associated disease.14 Furthermore, the two variants have been reported in the postpartum atypical hemolytic uremic syndrome, which was also due to the inappropriate activation of the complement alternative pathway.15–17 Thus, we inferred that these two variants might result in a mutant CFH protein with reduced regulatory capacity to prevent complement over-activation. Thrombomodulin is an endothelial anticoagulant glycoprotein that can regulate the complement alternative pathway by enhancing factor I-mediated C3b inactivation after binding C3b and CFH. Based on a study in a large cohort demonstrated that the THBD Ala473Val variant could increase the probability of developing C3G,12 we speculated that the mutant protein might reduce the binding capacity of C3b and CFH to inactive C3b, although the functional studies on THBD Ala473Val were lacking. In general, the common variants CFH Val62Ile, CFH Tyr402His and THBD Ala473Val found in our patient might cause the dysregulation of the complement alternative pathway and increase susceptibility to C3GN.
Another CFH variant Glu936Asp (rs1065489) was previously demonstrated to be associated with host susceptibility to meningococcal disease in genome wide association studies.18,19 CFH has been shown to be exploited by N. meningitidis to escape host immune control.20 Binding of N. meningitides to CFH would protect the bacterium from complement-mediated damage. Thus, we thought that this variant would increase the binding ability between CFH and N. meningitides and then facilitate the escape of the bacteria from host immune responses. The MBL2 Gly54Asp variant (rs1800450) was found to decrease MBL2 protein stability and expression,21 which was supported by the lower plasma MBL level (342.89 ng/ml) in our patient that could be defined as partial MBL deficiency (50–1000ng/ml).22 MBL is a soluble pattern recognition receptor that initiates the activation of the complement MBL pathway after binding to carbohydrate, and its deficiency was reported to increase the risk of various infections such as sepsis.23 Therefore, the common variants CFH Glu936Asp and MBL2 Gly54Asp might help to explain our patient's past history of meningitis.
In addition, the novel variant C2 (His18Arg) found in our patient had never before been reported in any database (ESP, 1KGP and ExAC) or in the literature. However, prediction methods (SIFT, Polyphen2_HDIV and GERP++) indicated that this missense mutation His18Arg in C2 might be conserved, disease-causing and influence the protein function. C2 is a key component in the classical and lectin pathways, thereby providing defense against microbial infection and assisting in the removal of immune components. It can be cleaved and bind to C4b to form C3 convertase (C4b2a). We hypothesized that this mutation in C2 might enhance the protein activity or prolong the half-life of C3 convertase and lead to the abnormal activation of MBL and/or the classical pathway. Considering that the patient's plasma C4d level was significant elevated and C1q was normal, we speculated that besides the alternative pathway, the MBL pathway might also be involved in the progression of C3GN in this patient.24 In addition, we were unable to know if this C2 mutation was inherited from his parents or not since they were unwilling to provide blood (DNA) samples. Nevertheless, it was still reasonable that they were not affected because of the incomplete penetrance, which means some individuals fail to express the related disease phenotype even though they carry the particular disease-causing mutation.25 The genetic background and gene-environment interplay are two of the underlying mechanisms of incomplete penetrance. Genetic background, such as the common variants in the CFH, THBD and MBL genes carried by this patient could largely predispose him to the development of C3GN. On the other hand, the environment will often influence clinical penetrance. The common environmental modifiers include diet, drugs, alcohol intake, metabolic syndromes and physical activity.26 Thus, the alcohol intake and/or any other unhealthy behaviors might exacerbate the impact of heritable genetic variants in this patient.
Therefore, it is logical to assume that, in this patient, common variants that could increase the risk of developing complement-mediated diseases (defined as susceptibility variants) combined with novel variants (defined as mutation) and a triggering event (such as infection) might lead to the development of an overt clinical and pathological phenotype. This finding supports that mutation alone in C3G might not be enough to increase its incidence risk, except when combined with common susceptibility variants and triggers.
The renal prognosis of C3G was poor, and previous reports showed that approximately 50% of DDD patients progressed to ESRD in 10 years.27 C3GN had a better prognosis than DDD, although the latest study by Bomback et al. showed that there was no significant difference in renal prognosis between DDD and C3GN.8 Unfortunately, there were still no high-level randomized trials for the treatment of C3GN, and the therapy for the disease was mainly based upon low-quality evidence consisting of case series, case reports, retrospective cohort studies and expert opinion.8,28 General treatments include antihypertensive therapy and lipid lowering therapy. Specific interventions include plasma infusion or exchange, immunosuppressive therapy and eculizumab.4 Since our patient had genetic defects of circulating proteins without acquired autoantibodies, he received fresh frozen plasma infusion, and immunosuppressants were not initiated. He responded well with an elevated plasma C3 level, well-controlled proteinuria and stable renal function. Moreover, anticomplement therapy (eculizumab) was a novel and pathogenesis-based treatment method for the disease in recent years. C3G patients with deteriorating renal function, severe nephrotic syndrome, elevated sMAC levels and active renal biopsy changes, might be more likely to benefit from this therapy based on previous studies,29,30 although the drug has not been approved for treatment of the disease in China.
ConclusionThis case highlights the importance of the combination of genetic mutation with susceptibility variants and triggers in the development of C3GN. Additional functional studies on these polymorphisms are required to better understand the pathophysiology of this disease.
Ethics approval and consent to participateThe research was in compliance with the Declaration of Helsinki. The design of this work was approved by the Medical Ethics Committee of Peking University First Hospital.
Consent for publicationWritten informed consent was obtained from the patient for publication of this case report.
Conflict of interestThe authors declare no conflicts of interest.
This work was supported by grants from the National Natural Science Foundation of China to the Innovation Research Group (No. 81621092) and the National Natural Science Foundation of China (No. 81470932, No. 81500526, No. 81500543, No. 81670640 and No., 81670639).



