









La acidosis tubular renal distal (ATRd) primaria es una tubulopatía poco frecuente caracterizada por la presencia de acidosis metabólica hiperclorémica. Está generada por la existencia de un defecto en la función de la H+-ATPasa situada en el lado luminal de las células α-intercaladas o del intercambiador de aniones Cl−-HCO3− (AE1) ubicado en el lado basolateral. Los pacientes no acidifican la orina tras una sobrecarga ácida (NH4Cl) o tras estimular la secreción de H+ mediante la obtención de una elevada concentración intratubular de un anión como cloro (se mide el pH) o HCO3− (se mide la pCO2 urinaria). Se presenta una familia con ATRd autosómica dominante producida por una mutación heterocigota en el gen SLC4A1 en la que los 2 miembros en edad pediátrica mostraron una prueba de la pCO2 urinaria máxima normal. Nuestra hipótesis es que al estar intacta, al menos inicialmente, la H+-ATPasa, podría ser efectivo el estímulo inducido por la electronegatividad intratubular para secretar H+ lo que permitiría que la pCO2 urinaria máxima fuera paradójicamente normal, lo que pudiera explicar el inicio tardío, la presentación moderada de los síntomas y el diagnóstico en edades más avanzadas, en los pacientes con dicha mutación. Este es el primer caso documentado de una ATRd dominante en México.
Primary distal renal tubular acidosis (dRTA) is a rare tubulopathy characterized by the presence of hyperchloremic metabolic acidosis. It is caused by the existence of a defect in the function of the H+-ATPase located on the luminal side of the α-intercalated cells or the Cl− HCO3− (AE1) anion exchanger located on the basolateral side. Patients do not acidify the urine after acid overload (NH4Cl) or after stimulating H+ secretion by obtaining a high intratubular concentration of an anion such as chlorine (pH is measured) or HCO3− (urinary pCO2 is measured). We present a family with autosomal dominant dRTA produced by a heterozygous mutation in the SLC4A1 gene in which the two pediatric members showed a test of normal maximum urinary pCO2. Our hypothesis is that since the H+-ATPase is intact, at least initially, the stimulation induced by intratubular electronegativity to secrete H+ could be effective, which would allow the maximum urinary pCO2 to be paradoxically normal, which could explain the onset, moderate presentation of symptoms and late diagnosis in patients with this mutation. This is the first documented case of a dominant dRTA in Mexico.
La acidosis tubular renal distal (ATRd) (enfermedad de Butler-Albright, nephrocalcinosis infantum) es un síndrome clínico caracterizado por la presencia de acidosis metabólica hiperclorémica que está causado por un defecto en la excreción urinaria de ion hidrógeno (H+) en las porciones distales de la nefrona y, por tanto, con incapacidad para disminuir el pH de la orina por debajo de 61,2. Es una enfermedad rara, que se manifiesta por retraso de crecimiento, poliuria, hipercalciuria, hipocitraturia, nefrocalcinosis, depleción de potasio y manifestaciones extrarrenales características en algunos de los tipos2,3.
La acidificación tubular distal de la orina en condiciones fisiológicas se resume en la figura 1. La H+-ATPasa (V-ATPasa) es una bomba de protones altamente conservada que se expresa en las células α-intercaladas y que está formada por dos dominios, V1 y V0. Los defectos en la actividad de la bomba H+-ATPasa causan la mayoría de los casos primarios de ATRd de herencia autosómica recesiva. Son debidos a mutaciones en los genes ATP6V1B1,ATP6V0A4 (tabla 1) que codifican las subunidades B1y A4 de la bomba H+ ATPasa, respectivamente4–6. La ATRd de herencia autosómica recesiva asociada a sordera puede ser producida por mutaciones en el gen FOXI 1 que codifica un factor de transcripción que es necesario para la expresión de al menos 4 subunidades de la H+-ATPasa (A1, B1, E2 y a4)7. Un cuarto gen, WDR72 (OMIM 613211), cuando está mutado produce otra variedad de ATRd autosómica recesiva; este gen parece estar implicado en el tráfico intracelular de proteínas reguladoras del equilibrio ácido-base provocando su retención intracelular o un direccionamiento erróneo8,9. Con relación a las manifestaciones extrarrenales: 1) los pacientes con ATRd recesiva causada por mutaciones en los genes ATP6V1B1, ATP6V0A4 pueden presentar una sordera de percepción de manera frecuente y constante en las mutaciones del gen FOXI 1, siendo de inicio temprano y asociado al síndrome de Pendred (ORPHA:705)8, y 2) los pacientes con ATRd recesiva causada por mutaciones del gen WDR72 pueden presentar una amelogénesis imperfecta.
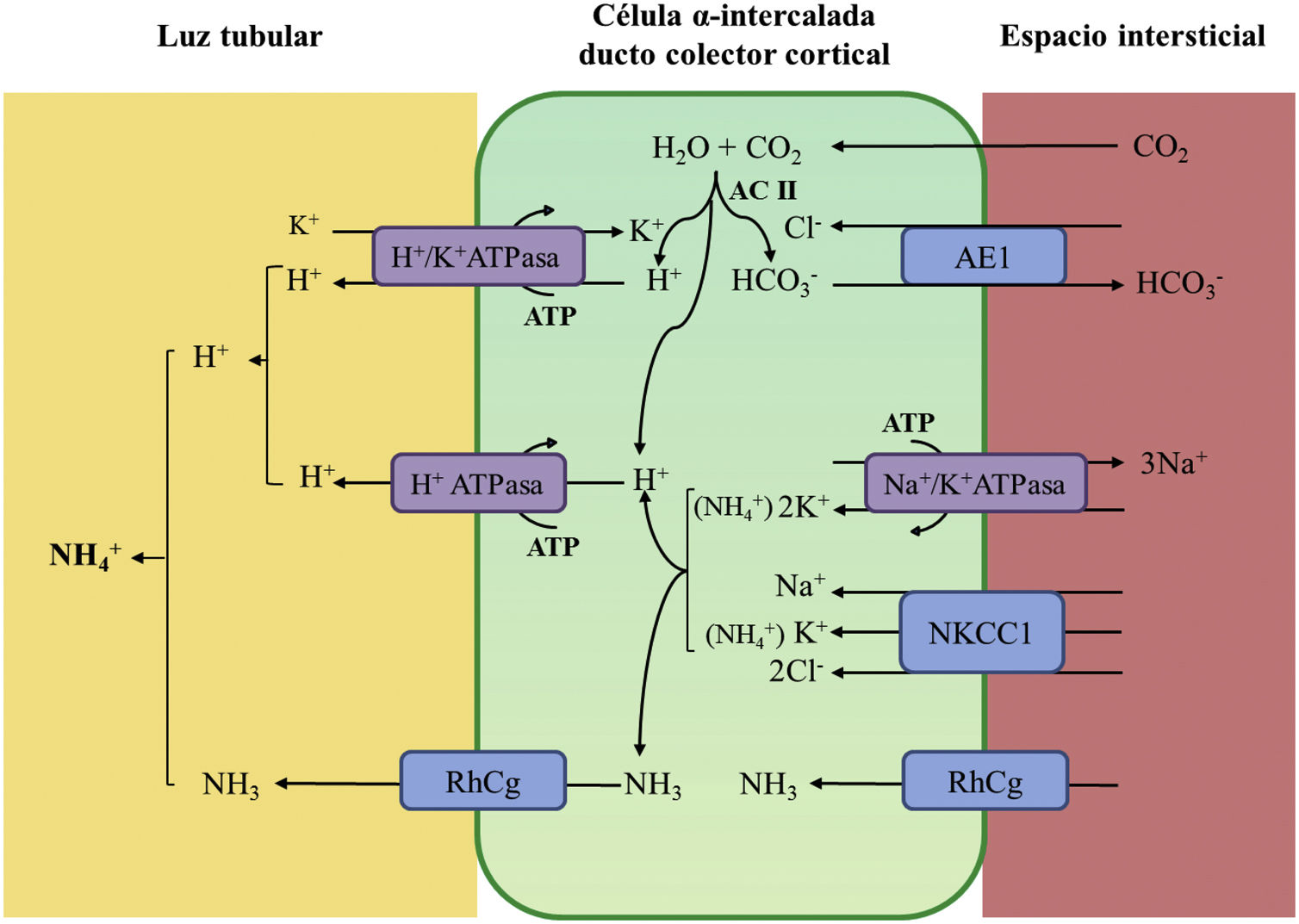
Acidificación de la orina distal en condiciones fisiológicas. La acidificación de la orina tiene lugar en los túbulos distal y colector a través de 3 procesos relacionados: a) reabsorción de la cantidad residual de bicarbonato (10%), que no ha sido recuperada en zonas más proximales de la nefrona; b) titulación del anión fosfato divalente (HPO42−) con H+ que se transforma en anión fosfato monovalente (H2PO4−) o acidez titulable; c) acumulación de amoniaco (NH3) intraluminal que capta H+ y forma amonio (NH4+). La secreción de H+ y la titulación de los tampones urinarios da lugar a la acidificación de la orina con lo que se pueden alcanzar valores de pH cercanos a 4,5 en condiciones de estimulación máxima del proceso. Este hecho se realiza en las células α-intercaladas que se localizan en el túbulo contorneado distal tardío, el túbulo conector y en los ductos colectores corticales y medulares.
La secreción distal de H+ genera una cantidad equimolar de bicarbonato. Por cada molécula de hidrogeno que se excreta a la luz tubular, se genera intracelularmente una molécula nueva de bicarbonato gracias a la acción de la anhidrasa carbónica intracitoplásmica (AC tipo II), que es transferida a la sangre mediante el intercambiador de aniones Cl−-HCO3− (AE1). Las células α-intercaladas secretan H+ por medio de la ATPasa vacuolar (H+-ATPasa) que transfiere H+ activamente a través de la membrana luminal y la H+-K+-ATPasa que intercambia H+ por potasio. La función de la H+-ATPasa está marcadamente influenciada por la electronegatividad generada en la luz tubular por el transporte simultáneo de Na+ en las células principales del ducto colector. La acumulación de NH3/NH4+ en la médula genera un gradiente de concentración que favorece su entrada por la membrana basolateral de las células intercaladas-α. La excreción de NH3/NH4+ requiere de al menos 2 pasos, a saber, ingreso basolateral y excreción luminal. La captación desde el intersticio del NH3/NH4+ se lleva a cabo por varias vías que incluyen el transportador Na+/K+/2Cl− (NKCC1), la Na+-K+-ATPasa (en el caso de estos dos trasportadores, el NH4+ puede ser transportado en lugar de potasio), los canales de gases Rhcg (human Rhesus C glycoprotein) y los canales de NH4+ activados por hiperpolarización HCN (hyperpolarization-activated cyclic nucleotide-gated cationic non-selective). La membrana luminal tiene una alta permeabilidad para NH3. Los canales RhCG están presentes en la membrana luminal y en la membrana basolateral.
OMIM Acidosis tubular renal distal hereditaria
| Gen | Proteína | EnfermedadOMIM | GeneOMIM | Transmsión |
|---|---|---|---|---|
| SLC4A1 | Intercambiador (AE1) | #179800 | *109270 | AD, AR |
| ATP6V1B1 | Subunidad B1 de la bomba H+ ATPasa (V-ATPasa) | #267300 | *192132 | AR |
| ATP6V0A4 | Subunidad a4 de la bomba H+ ATPasa (V-ATPasa) | #602722 | *605239 | AR |
En el lado basolateral solo se ha descrito un tipo de ATRd de herencia autosómico dominante (tabla 1) en la mayoría de los casos, que está producido por mutaciones en el gen SLC4A1 (OMIM 109270) que codifica las 2 isoformas del intercambiador de aniones Cl−-HCO3, la isoforma renal también conocida como kAE1 (kidneyanion exchanger 1) y la isoforma de los hematíes también conocida como proteína banda 3 (erythroid isoform o eAE1); kAE1 es responsable de la reabsorción de HCO3− junto con la excreción de Cl− en las células α-intercaladas10 (fig. 1). Este tipo de ATRd, se ha asociado a formas de presentación clínica menos graves, con inicio tardío en la infancia, adolescencia y en pacientes adultos, con menor impacto en el crecimiento comparado con las formas de herencia autosómico recesiva8.
Está descrito que los pacientes con ATRd no acidifican la orina tras una sobrecarga ácida (NH4Cl) o tras estimular la secreción de H+ mediante una elevada concentración intratubular conseguida a expensas de un anión como el cloro (furosemida, Cl2Ca). Tampoco elevan la pCO2 urinaria por encima de 70mmHg a pesar de obtenerse una elevada concentración intratubular de HCO3−11. Se presenta una familia con ATRd autosómica dominante producida por una mutación heterocigota en el gen SLC4A1 en la que dos de sus miembros en edad pediátrica mostraron una prueba de la pCO2 urinaria máxima normal.
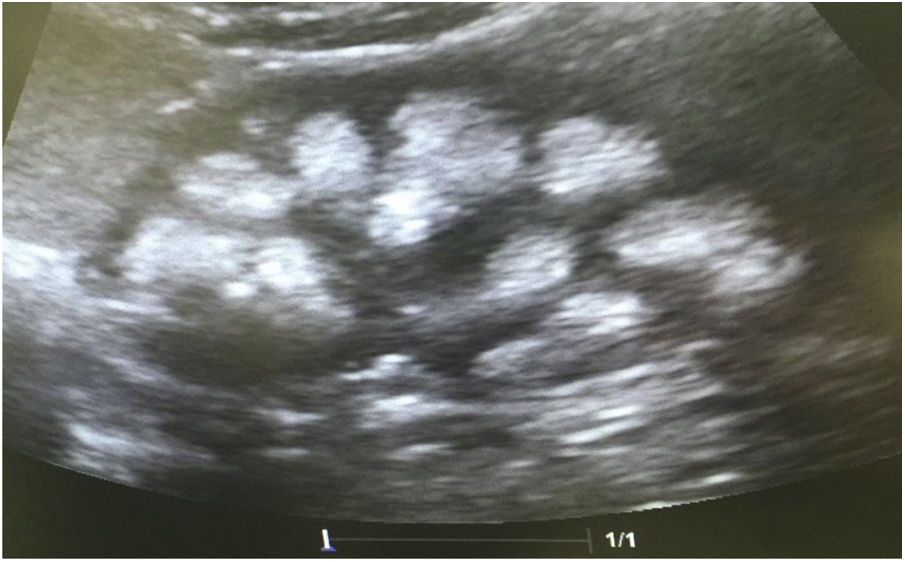
Casos clínicosCaso 1Paciente femenino de 4 años y 3 meses de edad. Peso al nacer 2.800g, talla 48cm y Apgar 8/9. Como antecedente presenta avidez por el agua y la sal. Poliuria y nicturia en la etapa de lactante. Fue hospitalizada en 11 ocasiones por fiebre sin foco aparente. A los 20 meses de edad fue diagnosticada de herpes zóster y tratada con aciclovir durante 7 días; a continuación, presentó un cuadro diarreico, deshidratación moderada y flacidez generalizada con incapacidad para caminar. Fue hospitalizada en el Hospital General de Zona con la sospecha de síndrome de Guillain-Barré; su peso era 9,5kg y la talla 82cm (ambos en percentil 10). Se observó hipopotasemia (2,5mEq/l) y acidosis metabólica. Tras instaurarse tratamiento intravenoso y la corrección del desequilibrio hidroelectrolítico, mejoró la fuerza muscular logrando la deambulación. Sin embargo, persistía la acidosis metabólica hiperclorémica. Los datos bioquímicos y los hallazgos de la ecografía aparecen en la tabla 2 y la figura 2, respectivamente. La valoración audiológica no mostró datos de hipoacusia neurosensorial. El estudio oftalmológico fue normal. Se realizó una prueba de acidificación urinaria con bicarbonato de sodio y acetazolamida según un protocolo descrito previamente (prueba de pCO2 urinaria máxima)11–13. La prueba se realizó sin complicaciones, fue bien tolerada y se consideró valida al conseguirse una bicarbonaturia superior a 80mEq/l. Se cuantificó una pCO2 urinaria máxima de 80mmHg a los 60min (anormal<70mmHg) (tabla 3). Se comprobó que la paciente es portadora de la misma mutación que su madre. Después de 3 años de tratamiento con álcalis ha mejorado el peso y la talla, a saber, 16,5kg (percentil 47) y 103cm (percentil 34), respectivamente. En la actualidad recibe tratamiento con citrato de potasio (4,5mEq/kg/día) con lo que ha remitido la hipercalciuria. La TA es normal (89/59mmHg).
Exámenes bioquímicos sanguíneos y urinarios
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
| Cistatina C (mg/l) | 0,91 | 0,99 | |
| FGRe (ml/min/1,73 m2)a | 102,03 | 92,12 | 122 |
| Gasometría venosa | |||
| pH | 7,33 | 7,04 | 7,17 |
| HCO3− (mEq/l) | 14,8 | 16,7 | 14,1 |
| Cloremia (mmol/l) | 112 | 114 | 113 |
| Potasemia (mmol/l) | 2,5 | 3,5 | 3,3 |
| Hiato aniónico sanguíneo | 11,2 | 8,3 | 10,9 |
| Uricemia (mg/dl) | 1,3 | 1,5 | 3,3 |
| Hiato aniónico urinario | 25,8 | 55,4 | 36 |
| Calciuria (mg/kg/día) | 10,5 | 8,9 | |
| Cociente calcio/creatinina en orina (mg/mg) | 0,95 | 0,84 | |
| Citraturia (mg/kg/día) | 0,5 | 0,42 | 0,24 |
| Cociente citrato/creatinina en orina (mg/g) | 0,413 | — | |
| Cociente calcio/citrato en orina (mg/mg) | 21 | 21,19 | |
| EF HCO3−(ml/100ml FGR) | 7 | 5,2 | |
| EFK (ml/100ml FGR) | 29,5 | 17 | |
| EF ácido úrico (ml/100ml FGR) | 16 | 13,9 | |
| Proteinuria (mg/m2/h) | 38 | 62 | |
EF: excreción fraccional; EFK: excreción fraccional de potasio; FGR: filtrado glomerular renal; FGRe: filtrado glomerular renal estimado; HCO3−: anión bicarbonato.
Paciente masculino de 8 años y 11 meses de edad sin antecedentes perinatales de importancia (peso 2,9kg, talla 50cm y Apgar 9/10). Avidez por el agua y la sal desde los 2 años de edad, aproximadamente. Precisó 3 hospitalizaciones durante su primer año de vida por episodios de deshidratación moderada. Acudió hasta en 24 ocasiones al servicio de urgencias de su localidad por la presencia de fiebre. En algunas ocasiones se catalogó de infección de vías respiratorias, si bien, en la mayoría de los casos no se detectó una causa aparente de la misma. Al efectuarse el diagnóstico de ATRd a su hermana, se realizó el abordaje diagnóstico. Su peso era 18kg (percentil 5) y la talla 111cm (percentil 3). Se observó acidosis metabólica hiperclorémica con anión restante normal, hipercalciuria, hipocitraturia severa (tabla 2) y nefrocalcinosis grado III. La valoración audiológica fue normal. La pCO2 urinaria máxima también fue normal (tabla 3). También, después de tres años de tratamiento con álcalis han mejorado tanto el peso (23kg, percentil 8) como la talla (122cm, percentil 17). Recibe tratamiento con citrato de potasio (5mEq/kg/día). El estudio genético mostró la misma mutación que su madre y su hermana.
En ambos niños la morfología de hematíes fue normal.
Caso 3La madre de los pacientes, de 29 años de edad, con antecedente de madre diabética y padre hipertenso en tratamiento. Hermano mayor con litiasis renal y episodios múltiples de parálisis flácida. Fue diagnosticada al realizar el abordaje diagnóstico de sus hijos. Con avidez por el agua desde la infancia. Su peso era 58kg, la talla 157cm y la presión arterial 100/60mmHg. Los niveles de creatinina eran 0,78mg/dl (CCr 122ml/min), el sodio 139mEq/l, el potasio 3,3mEq/l y la cloremia 113mEq/l. En la gasometría venosa se comprobó una acidosis metabólica (pH 7,17, HCO3− 14,1mEq/l) con hipocitraturia (14mg/24h) y proteinuria 382mg/24h (tabla 2). El pH urinario era 7 y el hiato aniónico (anión gap) urinario positivo (+36). En el ultrasonido renal se observó nefrocalcinosis medular grado II-III (fig. 3). Se comprobó la existencia de un cálculo en el tercio inferior ureteral derecho y otro de 1,5cm en la vejiga. Se realizó el estudio genético analizando los genes ATP6V0A4, ATP6V1B1 y SLC4A1. Se comprobó la presencia de una variante patogénica en heterocigosis en el exón 20 del gen SLC4A1, c.2710_*12, p. (Tyr904_Val911delins68) que se traduce en una deleción de 39 bases (27 del ultimo exón y 12 de la región 3’ UTR) que modifica la parte carboxi terminal de la proteína (deleción de los últimos 8 aminoácidos e inserción de una nueva secuencia de 68 aminoácidos). Esta variación no descrita en la literatura y no incluida en la base de datos, es considerada patógena, de acuerdo a la ACMG 2015, clase 5 (American College of Medical Genetics and Genomics); los criterios usados para clasificarla en clase 5 son: PVS1, PM2, PM1, PM4. Esta variación no está presente en la base de datos de población general gnomAD (criterio PM2)
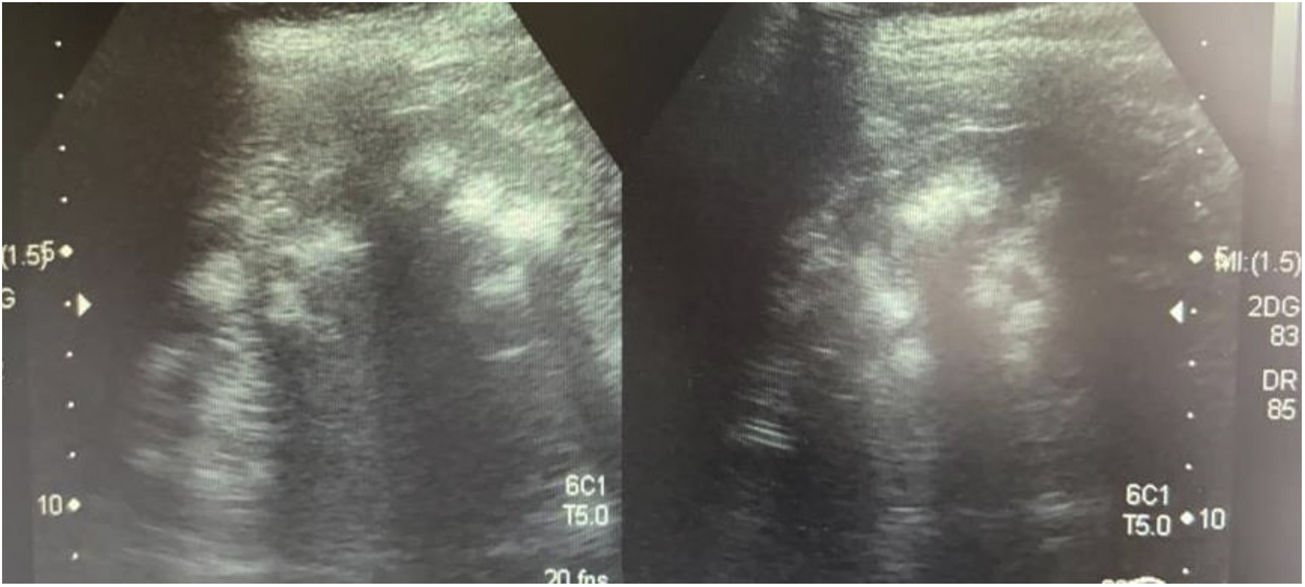
Ecografía realizada a la madre de los pacientes. Riñones con ecogenicidad heterogénea a expensas de un aumento de la ecogenicidad de las pirámides renales compatible con nefrocalcinosis medular bilateral grado II-III. La relación córtico medular es poco nítida, con datos de nefropatía crónica difusa.
Las muestras de sangre periféricas fueron recogidas en los tubos de EDTA. El ADN puro se obtuvo utilizando el kit MIDI de sangre QIAamp® ADN (Qiagen) según el fabricante. El caso índice de esta familia se analizó con el panel de genes usando «next generation sequencing» (descrito previamente en Ashton E et al. Kidney Int, 2018 PMID: 29398133 y Hureaux M Kidney Int, 2019 PMID: 31672324) que permitió el análisis de los 3 genes mayores; el FOXI1 no fue analizado, ya que no estaba incluido en el panel en el momento en que se realizó el estudio. Una vez identificada la variación se confirmó en el caso índice utilizando el método Sanger y se analizaron los otros miembros de la familia igualmente con Sanger. Los transcritos utilizados para describir las variaciones fueron: ATP6V0A4: NM_130841.2, ATP6V1B1: NM_001692.3 y SLC4A1: NM_000342.3
DiscusiónCuando existe un defecto genético en la actividad de la bomba H+-ATPasa, la acidosis metabólica causada por el defecto en la secreción tubular de H+ produce retraso del crecimiento, movilización del calcio óseo14 e inhibición de la reabsorción tubular de NaCl y calcio15. El citrato es un marcador muy sensible de acidosis metabólica16. Cuando el pH de la célula tubular proximal se reduce, se estimula la actividad del cotransportador de dicarboxilatos NADC1 (sodium-dependent dicarboxylate transporter 1) localizado en la membrana apical del túbulo proximal. El NADC1 es un transportador electrogénico acoplado a la reabsorción de Na+ (3 cationes Na+ por cada anión de cit2−). La consecuencia de una mayor reabsorción tubular es la hipocitraturia. Una vez reabsorbido el citrato, es transportado a las mitocondrias en las que la aconitasa mitocondrial lo transforma en isocitrato para entrar en el ciclo de Krebs17 en el que el citrato se convierte en CO2 y H2O y se consumen tres iones H+, una reacción equivalente a generar bicarbonato. La hipercalciuria y la hipocitraturia favorecen la aparición de nefrocalcinosis. Al no poder intercambiarse el Na+ por H+ en las porciones distales de la nefrona, necesita hacerlo con K+. La hipopotasemia se favorece por el hiperaldosteronismo secundario causado por la contracción de volumen que, a su vez, es originada por la pérdida salina y la poliuria. Ésta última, es generada por la nefrocalcinosis, la pérdida salina antes mencionada, la depleción corporal de potasio (nefropatía hipopotasémica)18,19 y, seguramente, por la propia hipercalciuria.
¿Qué ocurre cuando el defecto genético repercute en la actividad del intercambiador de aniones Cl−-HCO3− como es lo que le acontece a nuestros pacientes? El bicarbonato, formado intracelularmente por la acción de la anhidrasa carbónica no puede abandonar la célula a través de la membrana basolateral por lo que se eleva el pH intracelular que va a inhibir la actividad de la H+-ATPasa. Por ello, los pacientes con ATRd autosómica dominante tienen orinas con un pH superior a 6 en situación de acidosis espontánea20 o en las pruebas de acidificación realizadas con estímulo de H+ como en las efectuadas con NH4CL20,21. La falta de intercambio con Cl− explica la hipercloremia propia de la ATRd (fig. 1).
No obstante, al estar intacta, al menos inicialmente, la H+-ATPasa, podría ser eficaz el estímulo inducido por la electronegatividad intratubular. Desde los años 80 se reconoce que la capacidad de acidificación distal es dependiente de aniones22 y es estimulada por la aldosterona23. Este hecho explica, al menos, dos detalles importantes. En primer lugar, que los pacientes con la forma autosómica dominante se manifiesten clínicamente más tarde, sobretodo en la infancia, y en la adolescencia y comúnmente, como hallazgo causal en los adultos; con un fenotipo más leve en la adolescencia o en la edad adulta, con niveles de bicarbonato sérico ligeramente bajos y con menor afección y compromiso del peso y talla, las cuales suelen mantenerse en percentiles adecuados para la edad3,24. Incluso, algunos pacientes portadores de mutaciones heterocigotas en el gen SLC4A1 tienen un fenotipo de ATRd incompleta, es decir, sin acidosis metabólica20,25,26 (en algunos pacientes con ATRd incompleta la prueba de la pCO2 es normal25). En la evolución, seguramente se altera progresivamente la transcripción de las distintas subunidades integrantes de la H+-ATPasa con lo que la sintomatología se hace más evidente. En segundo lugar, la electronegatividad originada por la elevada concentración intratubular de bicarbonato estimularía a la H+-ATPasa a secretar H+ lo que permitiría que la pCO2 urinaria máxima fuera normal, como lo observado en los dos pacientes estudiados por nosotros.
Otros hallazgos importantes en los 3 casos presentados, es la hipouricemia, asociada a hiperuricosuria, que han sido descritos sobre todo en las formas autosómicas recesivas en algunos casos de ATRd, como consecuencia de la disfunción endosomal de las células del túbulo proximal inducida por la acidosis intracelular crónica o bien asociado a la nefropatía secundaria a la hipokalemia crónica, que condiciona la vacuolización de las células del túbulo proximal27,28.
Llama la atención la nefrocalcinosis tan importante que presentan los pacientes, que se ha descrito hasta en el 94% de los casos de ATRd con mutaciones en el gen SLC4A1, siendo discretamente más frecuente, hasta en un 98%, en las mutaciones ATP6V0A424, condicionada muy probablemente por la hipocitraturia severa en los 3 casos descritos, así como la hipercalciuria elevada que presentaban al ser diagnosticados, la cual se normalizó una vez que fue iniciado el tratamiento con citratos de potasio.
La herencia de este tipo de ATRd con mutaciones en el gen SLC4A1 es compleja, con una forma de transmisión, como ya se ha indicado, autosómica dominante, sobre todo, en caucásicos21. No obstante, también se ha descrito una forma recesiva, que es más común en algunos países tropicales del sudeste asiático, en particular en Tailandia, Malasia, Filipinas y Papúa Nueva Guinea, en estos países, la ovalocitosis debida a mutaciones heterocigotas de SLC4A1 (southeast Asian ovalocytosis [SAO]) es frecuente29,30. Otras mutaciones en este gen pueden causar igualmente esferocitosis. Las mutaciones que que causan la ovalocitosis o la esferocitosis son responsables de cambios morfológicos en los glóbulos rojos y de una anemia hemolítica y afectan aminoácidos diferentes de los que están afectados en la ATRd. Wrong et al.31 y otros grupos33 intentaron dilucidar la razón de la alta frecuencia de ATRd en el sudeste asiático. Fruto de ello fue una publicación en la que se sugería la hipótesis de que los cambios en los hematíes causados por estas mutaciones podrían proteger contra la malaria. En 1981, se demostró que los hematíes ovalocitos de los melanesios que habitan en Papúa Nueva Guinea son resistentes a la infección por parásitos de la malaria (Plasmodium falciparum)33. La esferocitosis hereditaria es una entidad fenotípica y genéticamente heterogénea. Entre el 20 y el 35% de los casos, son producidos por mutaciones en el gen SLC4A134. En este sentido, se han descrito pacientes con mutaciones bialélicas en ese gen (por ejemplo, una mutación de SAO y una de acidosis distal) que pueden padecer una anemia hemolítica asociada a ATRd35–37.
En 1999, Kaitwatcharachai et al. publicaron los datos de un paciente adulto con SAO en el que la prueba de la pCO2 urinaria fue normal38. Años después, uno de los coautores de este trabajo reflexionaba sobre este hallazgo e indicaba que alguna de proteínas kAE1 mutadas se expresaría en la membrana luminal lo que produciría «una secreción neta de bicarbonato y un aumento de la pCO2 en la nefrona distal»39. A nuestro modo de entender, esta hipótesis no explicaría el aumento de secreción de H+ que es necesaria para incrementar la pCO2 urinaria.
En todo caso, la mutación heterocigota en el exón 20 del gen SLC4A1 observada en nuestros pacientes es la primera vez que se describe. Por tanto, la normalización de la pCO2 urinaria observada en nuestros pacientes no debe ser específica de los pacientes portadores de la mutación SAO.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


AL DÍA


- Inicio
- Todos los contenidos
- Publique su artículo
- Acerca de la revista
- Métricas
- Open access