



La deficiencia de lecitina colesterol aciltransferasa (LCAT) es una enfermedad genética rara de herencia autosómica recesiva, causada por mutaciones en el gen LCAT que alteran el metabolismo de las lipoproteínas. Esto ocasiona una reducción marcada del colesterol HDL y acumulación de lipoproteína X, con daño renal, corneal y hemolítico. Existen dos variantes clínicas: deficiencia familiar completa (FLD) y síndrome de ojo de pez (FED).
Reportamos el caso de un varón de 41 años con antecedentes de hipertensión, tinnitus e hipoacusia progresiva, que desde la infancia presentó opacidad corneal bilateral. Se documentó proteinuria significativa (2,56g/24h), función renal conservada (creatinina 0,85mg/dL), anemia leve (Hb 10,2g/dL) y colesterol HDL extremadamente bajo (1,3mg/dL). La biopsia renal mostró glomeruloesclerosis focal y segmentaria y expansión mesangial; la microscopía electrónica reveló inclusiones laminares concéntricas conocidas como «cuerpos de cebra», hallazgo habitualmente descrito en la enfermedad de Fabry. No obstante, la actividad de alfa-galactosidasa A fue normal y el estudio genético para Fabry resultó negativo. El análisis genético adicional identificó la variante c.757 p.(Gln253Argfs*11) en el gen LCAT, confirmando deficiencia familiar de LCAT.
Este caso resalta la importancia del diagnóstico diferencial entre deficiencia de LCAT y enfermedad de Fabry, ya que comparten características clínicas e histológicas. Asimismo, constituye el primer reporte de «cuerpos de cebra» en deficiencia de LCAT, subrayando la complejidad diagnóstica y la necesidad de un abordaje multidisciplinario para optimizar el manejo de estos pacientes.
Lecithin–cholesterol acyltransferase (LCAT) deficiency is a rare autosomal recessive disorder resulting from mutations in the LCAT gene, which leads to abnormal lipoprotein metabolism. This results in markedly reduced high-density lipoprotein cholesterol and the accumulation of lipoprotein X, leading to renal, corneal, and hemolytic damage. Two clinical variants have been described: familial LCAT deficiency (FLD) and fish-eye disease (FED).
We report the case of a 41-year-old male with a history of hypertension, tinnitus, and progressive hearing loss, who presented with bilateral corneal opacity since childhood. Laboratory studies revealed significant proteinuria (2.56g/24h), preserved renal function (creatinine 0.85mg/dl), mild anemia (Hb 10.2g/dl), and extremely low HDL cholesterol (1.3mg/dl). Renal biopsy showed focal segmental glomerulosclerosis and mesangial expansion. Electron microscopy demonstrated concentric lamellar inclusions known as “zebra bodies,” a finding typically associated with Fabry disease. However, α-galactosidase A activity was normal, and genetic testing for Fabry disease was negative. Further genetic analysis identified the variant c.757 p.(Gln253Argfs*11) in the LCAT gene, confirming the diagnosis of familial LCAT deficiency.
This case highlights the importance of differentiating LCAT deficiency from Fabry disease, given their overlapping clinical and histological features. Moreover, it represents the first description of “zebra bodies” in LCAT deficiency, emphasizing the diagnostic complexity and the need for a multidisciplinary approach to ensure accurate diagnosis and appropriate management.
La deficiencia de lecitina colesterol aciltransferasa (LCAT) es una enfermedad genética rara, prevalencia menor 1/ 1.000000, de herencia autosómica recesiva causada por mutaciones del gen LCAT ubicado en el brazo corto del cromosoma 16, dando como consecuencia una alteración del metabolismo de las lipoproteínas1–3. La enzima LCAT es fundamental en la esterificación del colesterol libre en la superficie de las lipoproteínas de alta densidad (HDL) encargada de su transporte desde los tejidos periféricos hacia el hígado para su metabolismo4.
Su deficiencia lleva a la disminución de los niveles de HDL, apoA-I y apoA-II, aumento de colesterol libre y apo E; acumulación de lipoproteína X, lipoproteína compuesta por colesterol libre que no pudo ser esterificado y fosfolípidos, considerada la responsable del daño en diversos tejidos, como el riñón, ojos, hígado, bazo, médula ósea y los eritrocitos3,5–8.
La enfermedad tiene dos formas de presentación: la deficiencia familiar completa de LCAT ([FLD] Familiar LCAT disease o enfermedad de Norum, OMIM#2459009), que afecta a múltiples sistemas y la deficiencia familiar parcial de LCAT, el síndrome de ojo de pez ([FED] Fish eyes disease, OMIM#13612010), que se manifiesta exclusivamente con opacidades corneales y alteraciones lipídicas1,4, sin mayor compromiso sistémico.
Las manifestaciones clínicas de esta patología incluyen opacidad corneal que suele manifestarse desde la infancia en pacientes con FLD y FED, ocasionando alteraciones visuales severas que con frecuencia requieren trasplante. A nivel hematológico, los eritrocitos presentan morfología en «célula diana» secundaria a alteraciones en la composición lipídica de la membrana, lo que incrementa su fragilidad y conduce a hemólisis y anemia. En el perfil lipídico destaca una marcada reducción del colesterol HDL (< 10mg/dL) y una tasa de esterificación del colesterol (TEC) menor al 60%. Este hallazgo es característico de FLD y permite diferenciarlo de FED11,12.
El compromiso renal, presente en más del 50% de los casos descritos, constituye una de las principales causas de morbimortalidad. Se manifiesta como proteinuria desde edades tempranas, atribuida a la acumulación de lipoproteína X (LpX) en el glomérulo8. La LpX, una lipoproteína compuesta por fosfolípidos (60%) y colesterol no esterificado (30%), se deposita en la matriz mesangial y en la membrana basal, promoviendo disfunción endotelial, expansión mesangial y daño podocitario. Estos mecanismos favorecen la aparición de proteinuria progresiva y glomeruloesclerosis. Los hallazgos histológicos incluyen depósitos lipídicos irregulares, granulares, electrodensos o electrolúcidos, distribuidos en mesangio, subepitelio, subendotelio y membrana basal13–15.
Un aspecto clave en el diagnóstico de esta enfermedad es su diferenciación de otras condiciones con presentaciones clínicas y hallazgos histológicos similares, como la enfermedad de Fabry7,16. La presencia de cuerpos de cebra, inclusiones laminares concéntricas observadas en la microscopía electrónica, es un hallazgo que se asocia tanto a diversas condiciones como trastornos por depósitos lisosomales, así como en condiciones inducidas por medicamentos7,17–21; sin embargo, son característicamente asociadas a la enfermedad de Fabry, una enfermedad ligada a la deficiencia de la enzima alfa galactosidasa A y que condiciona la aparición de depósitos lisosomales ligada al cromosoma X, que también afecta el riñón, el corazón, y el sistema nervioso22,23. Esta similitud puede llevar a confusiones diagnósticas y, por lo tanto, es esencial realizar una evaluación exhaustiva, incluyendo estudios genéticos y enzimáticos, para establecer un diagnóstico preciso.
En este contexto, presentamos el caso de un paciente con nefropatía por deficiencia de LCAT, en quien inicialmente se sospechó de enfermedad de Fabry debido a los hallazgos histológicos en la biopsia renal. Este caso destaca la importancia del diagnóstico diferencial en enfermedades raras y subraya la necesidad de un enfoque multidisciplinario para lograr un diagnóstico certero y un manejo clínico adecuado.
Reporte de casoSe presenta el caso de un paciente varón de 41 años con antecedentes de hipertensión arterial (HTA), tinnitus e hipoacusia progresiva de causa desconocida desde hace tres años. En un control médico rutinario realizado hace dos años, se detectó microalbuminuria, seguida por la aparición de orinas espumosas y edemas en las extremidades inferiores y manos. Durante su evaluación se nota opacidad corneal bilateral, desde la niñez referido por el paciente, anemia leve sin etiología clara y dislipidemia (HDL bajo). Es relevante destacar que su hermano presenta una opacidad corneal similar, aunque sin manifestaciones de enfermedad renal.
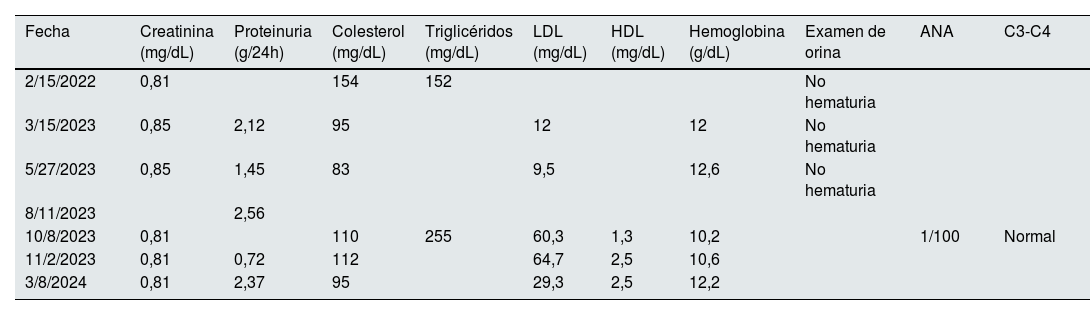
En agosto de 2023 es referido a la consulta nefrológica para estudio de proteinuria, se evidenció proteinuria significativa en rango no nefrótico (2,56g/24h), sedimento urinario sin presencia de hematuria o cilindros y función renal conservada, con niveles de creatinina sérica en 0,85mg/dL; además, hemoglobina en 10,2g/dL, colesterol en 110mg/dL, HDL: 1,3mg/dL y LDL: 60;3mg/dL, albumina sérica: 3,3g/dL triglicérido: 255mg/dL. Los resultados de anticuerpos antinucleares (ANCA), complemento sérico y virales fueron negativos (tabla 1). Debido a estos hallazgos, se decidió su hospitalización el 7 de octubre de 2023 para la realización de una biopsia renal.
Resultados de laboratorio, creatinina (creatinina sérica mg/dL), proteinuria (proteinuria 24 horas g/día), colesterol (colesterol sérico mg/dL), triglicéridos (triglicéridos séricos mg/dL), LDL (colesterol LDL mg/dL), HDL (colesterol HDL mg/dL), hemoglobina (hemoglobina g/dL), ANA (anticuerpos antinucleares patrón nuclear), C3-C4 (complemento sérico C3 y C4)
| Fecha | Creatinina (mg/dL) | Proteinuria (g/24h) | Colesterol (mg/dL) | Triglicéridos (mg/dL) | LDL (mg/dL) | HDL (mg/dL) | Hemoglobina (g/dL) | Examen de orina | ANA | C3-C4 |
|---|---|---|---|---|---|---|---|---|---|---|
| 2/15/2022 | 0,81 | 154 | 152 | No hematuria | ||||||
| 3/15/2023 | 0,85 | 2,12 | 95 | 12 | 12 | No hematuria | ||||
| 5/27/2023 | 0,85 | 1,45 | 83 | 9,5 | 12,6 | No hematuria | ||||
| 8/11/2023 | 2,56 | |||||||||
| 10/8/2023 | 0,81 | 110 | 255 | 60,3 | 1,3 | 10,2 | 1/100 | Normal | ||
| 11/2/2023 | 0,81 | 0,72 | 112 | 64,7 | 2,5 | 10,6 | ||||
| 3/8/2024 | 0,81 | 2,37 | 95 | 29,3 | 2,5 | 12,2 |
El examen de la microscopía óptica reveló 46 glomérulos, de los cuales siete mostraban esclerosis global. Los glomérulos viables presentaban expansión mesangial, rigidez de las asas capilares y esclerosis segmentaria en dos glomérulos, hallazgos compatibles con glomeruloesclerosis focal y segmentaria. La microscopía electrónica identificó inclusiones lisosomales laminares concéntricas, conocidas como «cuerpos de cebra», con una periodicidad de 3,95nm, localizadas en las asas capilares, mesangio, podocitos y miocitos de las arteriolas (fig. 1).
. Los podocitos viscerales y parietales, evidencian inclusiones lisosomales laminares concéntricas en menor grado que el endotelio.")
Microscopía electrónica. Se observan inclusiones lamelares tipo «cuerpos mielínicos», las asas capilares están completamente ocupadas por inclusiones lisosomales laminares concéntricas (cuerpos zebra). Los podocitos viscerales y parietales, evidencian inclusiones lisosomales laminares concéntricas en menor grado que el endotelio.
Ante estos hallazgos, se planteó la sospecha de enfermedad de Fabry, y se procedió a realizar estudios complementarios. La actividad enzimática de alfa galactosidasa A en sangre seca fue de 59,04 nmol/mg prot/h (rango de referencia: 20-100). El nivel de Lyso GL-3 fue <0,3 ng/dL y el estudio genético para la mutación en el gen GLA resultó negativo para enfermedad de Fabry.
La evaluación oftalmológica mostró opacidad en el estroma corneal periférico en ambos ojos, no compatible con córnea verticilata con lo cual se aleja el diagnóstico de enfermedad de Fabry desde el punto de vista oftalmológico (fig. 2). Dado el contexto clínico, se solicitó una evaluación genética adicional, la cual identificó la variante c.757 del p.(Gln253Argfs*11) en el gen LCAT, clasificada como variante de significado incierto según las directrices ACMG/AMP/ClinGen SVI implementadas por CENTOGENE. Estos hallazgos condujeron al diagnóstico de deficiencia de LCAT, una condición hereditaria de carácter autosómico recesivo.

El diagnóstico definitivo es deficiencia de LCAT en su variante familiar, sustentado en la presencia de niveles bajos de HDL, opacidad corneal, anemia y compromiso renal. El paciente ha sido manejado con Losartán 100mg/día y dapagliflozina 10mg/día, utilizados con el objetivo de disminuir la progresión de la proteinuria y el compromiso renal ante la falta de tratamiento específico disponible para esta patología.
DiscusiónLa deficiencia de LCAT es una enfermedad rara y de herencia autosómica recesiva que afecta el metabolismo de las lipoproteínas. Como se ha descrito en la literatura, las mutaciones en el gen LCAT comprometen la capacidad de la enzima para esterificar el colesterol libre, lo que resulta en una alteración importante del perfil lipídico y una acumulación de lipoproteína X, implicada en el daño a múltiples órganos, incluidos los riñones, los ojos y los eritrocitos1–6.
En este caso, el diagnóstico de deficiencia de LCAT se estableció tras una serie de evaluaciones clínicas, bioquímicas y genéticas, que mostraron un perfil lipídico alterado, niveles extremadamente bajos de HDL y una variante genética patogénica en el gen LCAT. En la literatura no se ha encontrado relación de alteraciones auditivas como las que presenta el paciente con esta patología. Las manifestaciones renales del paciente, que incluyen proteinuria significativa, son consistentes con lo reportado en otros estudios de esta rara enfermedad, sin embargo, el paciente mantiene buena función renal, por lo que el abordaje terapéutico temprano estaría orientado a disminuir la progresión de la enfermedad renal crónica.
Un hallazgo particularmente relevante en este caso fue la identificación de cuerpos de cebra en la microscopía electrónica, un tipo de inclusión laminar concéntrica que se asocia clásicamente con la enfermedad de Fabry. Sin embargo, este tipo de hallazgo no había sido previamente descrito en la deficiencia de LCAT, lo que subraya su importancia y destaca la complejidad del diagnóstico diferencial en este paciente. La presencia de cuerpos de cebra inicialmente planteó la sospecha de enfermedad de Fabry, una condición de depósito lisosomal que también afecta el riñón, el corazón y otros órganos. Este hallazgo novedoso puede sugerir que las inclusiones laminares no son exclusivas de la enfermedad de Fabry, y que la deficiencia de LCAT puede compartir algunas características histológicas similares, lo cual requiere más investigación.
El diagnóstico diferencial con la enfermedad de Fabry fue resuelto mediante estudios genéticos que confirmaron la ausencia de mutaciones en el gen GLA, así como a través de la medición de la actividad enzimática de alfa galactosidasa A, que resultó normal. Estos hallazgos orientaron hacia el diagnóstico definitivo de deficiencia de LCAT.
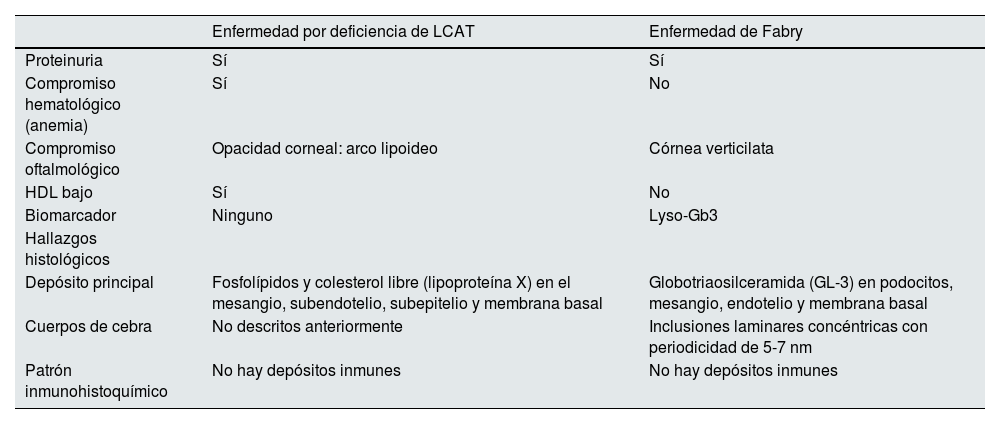
Es importante señalar que el cuadro clínico de la enfermedad de Fabry y la deficiencia de LCAT, muestran diferencias que pueden orientar el diagnóstico diferencial (tabla 2).
Cuadro comparativo de características clínicas y anatomopatológicas de la Enfermedad por deficiencia de LCAT y enfermedad de Fabry
| Enfermedad por deficiencia de LCAT | Enfermedad de Fabry | |
|---|---|---|
| Proteinuria | Sí | Sí |
| Compromiso hematológico (anemia) | Sí | No |
| Compromiso oftalmológico | Opacidad corneal: arco lipoideo | Córnea verticilata |
| HDL bajo | Sí | No |
| Biomarcador | Ninguno | Lyso-Gb3 |
| Hallazgos histológicos | ||
| Depósito principal | Fosfolípidos y colesterol libre (lipoproteína X) en el mesangio, subendotelio, subepitelio y membrana basal | Globotriaosilceramida (GL-3) en podocitos, mesangio, endotelio y membrana basal |
| Cuerpos de cebra | No descritos anteriormente | Inclusiones laminares concéntricas con periodicidad de 5-7 nm |
| Patrón inmunohistoquímico | No hay depósitos inmunes | No hay depósitos inmunes |
El manejo del paciente se centró en el control de la proteinuria y la hipertensión, utilizando losartán y dapagliflozina. Sin embargo, en la actualidad, no existe un tratamiento curativo para la deficiencia de LCAT, y las intervenciones terapéuticas están enfocadas en el manejo sintomático de las complicaciones14. La administración de LCAT recombinante se ha estudiado como una posible terapia para corregir la dislipidemia en estos pacientes, pero aún no está ampliamente disponible14,24,25. En este caso, el paciente ha sido manejado con terapia conservadora, y se requiere un seguimiento a largo plazo para monitorear la progresión de la insuficiencia renal y evaluar la necesidad de terapia de reemplazo renal.
Este caso subraya la importancia de un enfoque multidisciplinario en el diagnóstico y manejo de la deficiencia de LCAT, una enfermedad rara pero clínicamente significativa. Además, resalta la relevancia del hallazgo de cuerpos de cebra en la microscopía electrónica, que no había sido descrito previamente en esta condición, enfatizando la necesidad de una evaluación exhaustiva para diferenciar esta patología de otras con características histológicas similares como la enfermedad de Fabry.
Durante la preparación de este manuscrito los autores utilizaron ChatGPT a fin de mejorar la redacción. Tras la utilización de esta herramienta de Inteligencia Artificial, realizamos una revisión y edición del contenido, asumiendo la plena responsabilidad del contenido de la publicación.
Identificadores ORCIDJuan Enrique Rodriguez Mori: 0000-0002-9794-0327
Milagros del Pilar Dapello Jimenez: 0009-0004-2072-525X
Julia Sumire Umeres: 0000-0002-9505-3783
Roxana Maybor Lipa Chancolla: 0000-0002-8653-8906
FinanciaciónEl artículo ha sido autofinanciado por el equipo de investigación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



