


Gitelman syndrome (GS) is one of the most frequent inherited renal tubular disorders,1 and it's caused by mutations in the SLC12A3 gene encoding the thiazide-sensitive sodium chloride cotransporter (NCC) expressed in the apical membrane of distal convoluted tubule (DCT) cells.2
We present the case of a 50-year-old caucasian male with persistent hypokalaemia, referred to our nephrology department. This condition had first been found in blood analyses made on an emergency room visit due to a syncope and, until then, monitored in the primary care. In the anamnesis he only refers occasional paresthesias on the lower limbs, denying other symptoms. He has history of a left adrenal nodule (9mm) stable since 2011 and was on supplementation with 600mg of potassium chloride twice a day. His family history was negative. On physical examination he had a normal blood pressure, no significant alterations were found.
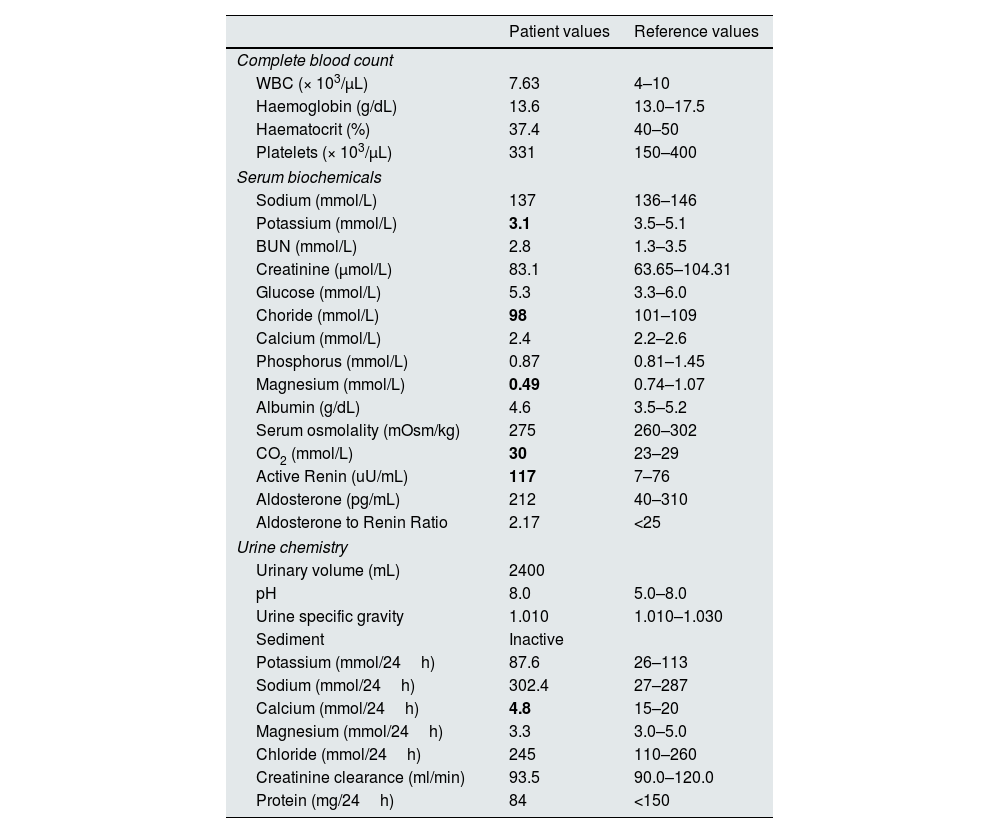
On biochemical analyses the patient presented persistent hypokalaemia, despite supplementation, hypomagnesaemia and hypochloremia. Serum creatinine, and remainder ionogram were normal. Further investigation revealed elevated plasma-active renin, normal aldosterone, elevated CO2 and hypocalciuria (Table 1).
Laboratory investigation.
| Patient values | Reference values | |
|---|---|---|
| Complete blood count | ||
| WBC (× 103/μL) | 7.63 | 4–10 |
| Haemoglobin (g/dL) | 13.6 | 13.0–17.5 |
| Haematocrit (%) | 37.4 | 40–50 |
| Platelets (× 103/μL) | 331 | 150–400 |
| Serum biochemicals | ||
| Sodium (mmol/L) | 137 | 136–146 |
| Potassium (mmol/L) | 3.1 | 3.5–5.1 |
| BUN (mmol/L) | 2.8 | 1.3–3.5 |
| Creatinine (μmol/L) | 83.1 | 63.65–104.31 |
| Glucose (mmol/L) | 5.3 | 3.3–6.0 |
| Choride (mmol/L) | 98 | 101–109 |
| Calcium (mmol/L) | 2.4 | 2.2–2.6 |
| Phosphorus (mmol/L) | 0.87 | 0.81–1.45 |
| Magnesium (mmol/L) | 0.49 | 0.74–1.07 |
| Albumin (g/dL) | 4.6 | 3.5–5.2 |
| Serum osmolality (mOsm/kg) | 275 | 260–302 |
| CO2 (mmol/L) | 30 | 23–29 |
| Active Renin (uU/mL) | 117 | 7–76 |
| Aldosterone (pg/mL) | 212 | 40–310 |
| Aldosterone to Renin Ratio | 2.17 | <25 |
| Urine chemistry | ||
| Urinary volume (mL) | 2400 | |
| pH | 8.0 | 5.0–8.0 |
| Urine specific gravity | 1.010 | 1.010–1.030 |
| Sediment | Inactive | |
| Potassium (mmol/24h) | 87.6 | 26–113 |
| Sodium (mmol/24h) | 302.4 | 27–287 |
| Calcium (mmol/24h) | 4.8 | 15–20 |
| Magnesium (mmol/24h) | 3.3 | 3.0–5.0 |
| Chloride (mmol/24h) | 245 | 110–260 |
| Creatinine clearance (ml/min) | 93.5 | 90.0–120.0 |
| Protein (mg/24h) | 84 | <150 |
Abdominal computer tomography revealed normal kidneys and no adrenal nodule was found. The patient started supplementation with magnesium and spironolactone.
Due to high suspicion of Gitelman syndrome, a genetic test was performed using next generation sequencing Ion AmpliSeq Exome Panel (Hi-Q) Kit. The test found an apparent homozygous mutation, c.945del p(Gly316Alafs*54), in the SLC12A3 gene. Sequence map is shown in Fig. 1.
 mutation in apparent homozygoty.")
In our first evaluation, we could think about primary hyperaldosteronism as a possible cause for the low serum potassium associated with a left adrenal nodule. However, the patient presented normal blood pressure and the CT was normal.
After the results from further investigation (Table 1) the main differential diagnosis were eating disorders, long-term laxative abuse, thiazide diuretics abuse, and Bartter and Gitelman syndromes.3 These non-renal causes were excluded by normal urinary chloride excretion, the absence of metabolic acidosis, consistent analysis results over time and normal urinary sodium and potassium excretion.3 Bartter syndrome was less likely as it is characterized by an earlier onset, associated with growth retardation. Low urinary calcium excretion excluded this tubulopathy.1 The final diagnose of GS was obtained with the genetic test.
First described by Gitelman et al. in 1966, GS is an autosomal recessive salt-losing renal tubulopathy,4 in most cases due to inactivating mutations in the gene that encodes the renal thiazide-sensitive NCC present in the epithelial cells of the DCT.2 With an estimated prevalence of ∼25 per million, GS is the most frequent inherited tubulopathy.1
It is characterized by hypomagnesaemia, hypocalciuria and secondary aldosteronism, responsible for hypokalaemia and metabolic alkalosis.4
Patients usually present above six years of age and in many cases the diagnosis is only made at adult age. Most suffer from tetany, especially during periods of fever or gastrointestinal losses, and paresthesias.5
GS is caused, in the majority of cases, by mutations in the solute carrier family 12, member 3, SLC12A3 gene.1 To date, >160 mutations, including missense, nonsense, frameshift, and splice-site mutations, as well as gene rearrangements, have been documented.6 Missense mutations are the most frequent, being the IVS9+1G>T reported as the most common mutation in the European population, and frameshift mutations are much fewer.7
GS diagnosis is based on the clinical symptoms and biochemical abnormalities above described,1 and confirmed by genetic test.8 Our patient presented an apparent homozygous mutation, in the SLC12A3 gene. This variant is found on the gnomAD data base, with an allelic frequency of 1:251374 in the general population, but it has never been identified in homozygoty or in European population, which highlights the relevance of this publication. This is a frameshift mutation on exon 7, which began from the glycine in the No. 316, mutated into alanine, and leading to premature termination of NCC protein, that affects the splicing process and introduces a premature stop-codon (Human Splicing Finder web source was used to predict the possible consequences of the mutation in the splicing process).9
The long-term treatment relies on high intake potassium and non-restrictive salt diet, as well as on supplements and other drugs. Lifelong oral potassium and magnesium supplementation are the mainstay of treatment for these patients.8 In cases of persistent or symptomatic hypokalaemia, the use of potassium-sparing diuretics can be useful, as they increase serum potassium levels and treat magnesium depletion that is worsened by elevated aldosterone levels.10
In summary, it would be interesting to study the first-degree relatives to exclude the possibility that the variant found is a heterozygous mutation with deletion of the other allele in this locus. Also, it is important to study the other family members to understand if there is an history of consanguinity that could explain the presence of this rare homozygous mutation and alert to the possibility of other genetic diseases yet to detect.
Conflict of interestNone.



