

El magnesio es el catión extracelular más abundante en el cuerpo humano y el segundo más abundante intracelular después del potasio. Es esencial para la transferencia, almacenamiento y utilización de la energía como regulador y catalizador de más de 300 sistemas enzimáticos. La hipomagnesemia puede producir una variedad de anormalidades metabólicas y consecuencias clínicas. Puede resultar del desequilibrio entre la absorción intestinal y la excreción renal. La principal consecuencia relacionada directamente con la hipomagnesemia son las arritmias cardiovasculares por hipopotasemia secundaria, y si no se reconoce y trata puede ser fatal. En este artículo revisamos las hipomagnesemias haciendo hincapié en los mecanismos moleculares responsables de la homeostasis del magnesio, diagnóstico diferencial y tratamiento, a propósito de la descripción de las manifestaciones clínicas y bioquímicas y el defecto genético en una familia afectada de síndrome de Gitelman.
INTRODUCCIÓN
El magnesio es el cuarto catión más abundante en el organismo y el segundo catión más abundante en el compartimiento intracelular. Es esencial para la función de muchas enzimas, incluyendo aquellas que se encuentran relacionadas con la transferencia de grupos fosfato, todas las reacciones que requieren ATP y cada paso relacionado con la replicación y trascripción del ADN y la traducción del ARNm. Se estima que el magnesio corporal total constituye unos 1.000 mmol o 22,66 g. El 99% del magnesio corporal total está localizado en el compartimiento intracelular. De dicho total, el 60% está localizado en el hueso, el 20% en el músculo y otro 20% en otros tejidos. La traslocación de magnesio del compartimiento intracelular al extracelular se realiza de manera lenta, durante varias semanas. Sólo el 1% del magnesio corporal total está ubicado en el compartimiento extracelular. De este total, el 60% se encuentra en forma libre o ionizada, el 10% está ligado a sales de citrato, fosfato, oxalato y otros aniones formando complejos y un 30% está ligado a proteínas. La concentración de magnesio en el plasma es mantenida en un estrecho rango comprendido entre 1,7 y 2,2 mg/dl (0,75-0,95 mmol/l o 1,5-1,9 mEq/l). La homeostasis del magnesio depende del equilibrio entre su absorción intestinal y su excreción renal. La hipomagnesemia se define como una concentración plasmática de magnesio menor de 1,7 mg/dl (<0,75 mmol/l o <1,5 mEq/l).
FISIOLOGÍA DE LA HOMEOSTASIS DEL MAGNESIO
En la dieta promedio se ingieren 360 mg (15 mmol) de magnesio elemental. El requerimiento diario de magnesio elemental es 0,15 a 0,20 mmol/kg. Entre las fuentes ricas en magnesio se incluyen los cereales, granos, nueces, legumbres, chocolate, vegetales verdes, y algunas carnes y mariscos. De forma habitual, sólo el 50% del magnesio de la dieta es absorbido en el tracto gastrointestinal, primariamente en el yeyuno proximal y el íleo. Alrededor de 40 mg/día de magnesio son también secretados en el intestino y, de ellos, sólo 20 mg son reabsorbidos en el colon y en el recto.
La absorción de magnesio en el íleo se produce mediante dos procesos:
El primero es un proceso activo y saturable, que constituye la ruta principal de transporte de magnesio. Éste se realiza a través del canal de magnesio TRPM61,2,3, (transient receptor potential melastatin). Un segundo mecanismo, que es pasivo y no saturable, se realiza a través de la ruta paracelular1,2.
El magnesio es esencial en la transferencia, almacenamiento y utilización de energía, regulando y catalizando más de 300 sistemas enzimáticos. La hipomagnesemia puede, por tanto, producir una variedad de anormalidades metabólicas y de consecuencias clínicas, que se ocasionan por un desequilibrio entre la absorción gastrointestinal y la excreción renal de magnesio. La principal manifestación de hipomagnesemia son las arritmias cardíacas que, de no ser reconocidas y tratadas, pueden ser fatales.
El 80% de magnesio en el plasma es filtrado por el glomérulo, del cual un 95% es reabsorbido por la nefrona. A diferencia de otros iones, la absorción tubular de magnesio ocurre sobre todo en el asa gruesa de Henle, siendo ésta de un 60 a un 70% del total filtrado. El túbulo proximal absorbe sólo un 15-25% del magnesio filtrado; por su parte, el túbulo distal absorbe un 5-10% del magnesio filtrado, pero se considera como el sitio de control final en la regulación de magnesio1. En el asa gruesa de Henle, el magnesio es reabsorbido con el calcio de manera pasiva a través de la vía paracelular formada por uniones intercelulares estrechas4-6. La fuerza que impulsa esta reabsorción es el gradiente eléctrico generado por la reabsorción de sodio a través del cotransportador Na+/K+/2Cl- (NKCC2). La paracelina-1, también conocida como claudina-16, ha sido identificada como la proteína constituyente de estas uniones intercelulares estrechas4-6.
En el túbulo distal, el magnesio es reabsorbido a través de un mecanismo activo, que implica al canal de magnesio TRPM67. El mecanismo de transporte del magnesio en la membrana basolateral de las células del asa gruesa de Henle y túbulo distal se desconoce. El transporte de magnesio en esta membrana debiera ser en contra de un gradiente electroquímico. La mayoría de los estudios apuntan hacia un mecanismo de intercambio dependiente del sodio, que se vería favorecido por bajas concentraciones de sodio intracelular generadas por la bomba de Na+/K+-ATPasa8.
FACTORES QUE INFLUYEN EN LA EXCRECIÓN DE MAGNESIO POR EL RIÑÓN
1. La concentración plasmática de magnesio es el principal regulador de la excreción de magnesio en el riñón. La hipermagnesemia inhibe la reabsorción de magnesio en el asa gruesa de Henle, mientras que la hipomagnesemia la estimula. La concentración plasmática de calcio posee un efecto similar. La hipermagnesemia y la hipercalcemia inhiben la reabsorción de magnesio a través de la activación del receptor-sensor del calcio de las células del asa gruesa de Henle y del túbulo distal. Cuando el magnesio o el calcio activan el receptor, se estimula la formación de un derivado del ácido araquidónico, el cual inhibe, de manera reversible, los canales de potasio en el asa gruesa de Henle. La secreción de potasio tiene dos funciones: en primer lugar, provee potasio para la reabsorción de sodio y cloro a través del cotransportador NKCC2 y, en segundo, interviene en la producción del gradiente eléctrico necesario para la reabsorción pasiva de magnesio y calcio9. Por tanto, la inhibición de los canales de potasio en el asa gruesa de Henle reduciría tanto el transporte de sodio como la reabsorción pasiva de magnesio y calcio10,11.
2. El volumen del fluido extracelular también influye sobre la excreción de magnesio. La expansión de volumen inhibe la reabsorción de magnesio en el asa gruesa de Henle, probablemente a causa de un incremento en la carga de sodio y, por ende, una disminución en el gradiente eléctrico, que favorece el transporte paracelular de magnesio.
3. Los cambios en la tasa de filtración glomerular también pueden influir en la excreción renal de magnesio. Cuando la tasa de filtración glomerular disminuye y, en consecuencia, la carga de magnesio filtrada, desciende la reabsorción de magnesio.
4. La depleción de fosfato disminuye la reabsorción de magnesio por un mecanismo desconocido.
5. La acidosis metabólica crónica produce una pérdida renal de magnesio, mientras que la alcalosis metabólica crónica causa el efecto opuesto. La acidosis metabólica crónica disminuye la expresión del canal de magnesio TRPM6 en el túbulo distal, disminuyendo la reabsorción de magnesio en esa localización. La alcalosis metabólica crónica aumenta la expresión de este canal, lo cual produce el efecto opuesto.
Varias hormonas, incluyendo la 1,25(OH)2 vitamina D, parathormona, calcitonina, glucagón, aldosterona, hormona antidiurética, insulina, prostaglandina E2 y catecolaminas aumentan la reabsorción de magnesio en el asa gruesa de Henle y el túbulo distal. El mecanismo se desconoce, pero se cree que en muchos casos esto estaría relacionado con el incremento de AMPc intracelular. Además, estudios recientes demuestran que la 1,25(OH)2 vitamina D3 produciría también un incremento en la expresión de paracelina-1 a través de la activación del factor de transcripción PPAR y su posterior unión al elemento de respuesta específico PPRE en la región promotora del gen hPCLN-1.
CAUSAS DE HIPOMAGNESEMIA
La hipomagnesemia se puede producir por cuatro mecanismos fisiopatológicos:
Disminución de la ingesta
La disminución en la ingesta rara vez causa deficiencia de magnesio, ya que la mayoría de alimentos contienen cantidades significativas de este elemento y el riñón es capaz de adaptarse y conservar magnesio de manera muy eficiente. Sin embargo, la hipomagnesemia puede ocurrir en tres grupos de pacientes: pacientes desnutridos, pacientes alcohólicos y pacientes a quienes se les administra nutrición parenteral total durante tiempos prolongados.
Redistribución
La traslocación de magnesio del extracelular al intracelular es una causa frecuente de hipomagnesemia. Esto puede ocurrir en el denominado síndrome del hueso hambriento, en el cual el magnesio se deposita en el hueso. Este síndrome se produce en pacientes con hiperparatiroidismo después de haber sido sometidos a una paratiroidectomía o en pacientes con hipertiroidismo después de una tiroidectomía.
La hipomagnesemia puede producirse también debido a hiperinsulinemia durante el tratamiento de la cetoacidosis diabética, en el síndrome de realimentación o durante la administración intravenosa de dextrosa.
Pérdida gastrointestinal
Alteraciones de la absorción del magnesio en el intestino pueden ocurrir como consecuencia de diarrea por cualquier causa o con motivo de una resección quirúrgica del intestino. Los pacientes con ileostomías pueden desarrollar hipomagnesemia porque se produce cierta reabsorción de magnesio en el colon.
La hipomagnesemia con hipocalcemia secundaria (HHS) es una alteración autosómica recesiva caracterizada por hipomagnesemia grave asociada con hipocalcemia. La fisiopatología de la hipomagnesemia en esta entidad está relacionada con un defecto en la reabsorción de magnesio en el intestino y en el túbulo distal. Recientemente, mutaciones en el gen TRPM6, que expresa el canal de magnesio TRPM6, se han identificado como la alteración genética subyacente8.
Pérdida renal
Varias alteraciones tubulares hereditarias son responsables de la pérdida urinaria de magnesio. El síndrome de Gitelman es un trastorno autosómico recesivo causado por mutaciones en el gen SCL12A3 que expresa el cotransportador NaCl (NCCT) en el túbulo distal. Este síndrome se caracteriza por hipopotasemia, hipomagnesemia e hipocalciuria asociadas con alcalosis metabólica. La hipomagnesemia se encuentra presente en la mayoría de los pacientes con síndrome de Gitelman y en el pasado se asumía que estaba relacionada con el defecto en el cotransportador NCCT, pero el mecanismo exacto se desconocía. Recientemente, algunos estudios apuntan a que la pérdida de magnesio se debería a la disminución en la expresión del canal de magnesio TRPM6 en el túbulo distal12-16.
De las cinco variantes del síndrome de Bartter, sólo el síndrome de Bartter clásico o tipo III está relacionado con la hipomagnesemia. Esta variante del síndrome de Bartter está causada por mutaciones en el gen CLCNKB, que expresa el canal de cloro CLC-Kb localizado en la membrana basolateral del asa gruesa de Henle y túbulo distal. Este canal media el flujo de cloro al intersticio. Se ignora el mecanismo de la hipomagnesemia en este síndrome.
MANIFESTACIONES CLÍNICAS DE HIPOMAGNESEMIA
La mayoría de pacientes con hipomagnesemia no presentan síntomas. Los síntomas de hipomagnesemia no aparecen hasta que la concentración de magnesio plasmática desciende a valores inferiores a 1,2 mg/dl. Además, la hipomagnesemia se presenta acompañada por otras alteraciones electrolíticas, como hipopotasemia e hipocalcemia, lo cual hace difícil distinguir las manifestaciones clínicas17-20 relacionadas solamente a la deficiencia de magnesio. La clínica de presentación del síndrome de Gitelman es muy heterogénea, con sintomatología tan sutil como mareos o vértigo, debilidad muscular, calambres y dolor muscular, dolor articular, fatiga o cansancio21-25.
La hipopotasemia es un hallazgo habitual en pacientes con hipomagnesemia, que se produce en el 40-60% de los casos. En parte, ésta se debe a la enfermedad subyacente que causa tanto pérdidas de magnesio como de potasio, lo que sucede, por ejemplo, en pacientes que toman diuréticos o en los que tienen diarrea. No obstante, en realidad, el principal mecanismo de hipopotasemia causada por hipomagnesemia tiene que ver con las propiedades biofísicas intrínsecas de los canales ROMK126 que median la secreción de potasio en el asa gruesa de Henle. Los canales ROMK1 son canales rectificadores internos de potasio, lo que significa que tienen mayor conductancia para el potasio que fluye hacia dentro de la célula que el que lo hace hacia fuera de la célula. El mecanismo de esta conductancia preferencial hacia dentro de la célula resulta de la unión y bloqueo subsiguiente de la conductancia de potasio hacia fuera de la célula por el magnesio intracelular y poliaminas. Una reducción en el magnesio intracelular producto de la deficiencia de magnesio produciría una disminución en la rectificación interna y, por ende, un incremento de la conductancia de potasio hacia fuera de la célula, con la consiguiente pérdida de potasio e hipopotasemia.
De cualquier modo, la hipopotasemia inducida por hipomagnesemia se caracteriza por ser refractaria al tratamiento con suplementos de potasio y sólo podrá ser corregida con la corrección de la deficiencia de magnesio.
La hipomagnesemia también puede inducir hipocalcemia. Esto ocurre generalmente cuando la hipomagnesemia es grave (<1,2 mg/dl). El mecanismo es múltiple. Hay una disminución de la liberación de parathormona. El mecanismo no se conoce con detalle, pero se cree que el magnesio incrementaría la actividad de la subunidad alfa de la proteína G relacionada con el CaSR. La hipomagnesemia también puede causar resistencia a las acciones de la parathormona en el tejido óseo. Al parecer, la deficiencia de magnesio interfiere con la generación de AMPc, que es el mediador intracelular de la parathormona.
ESTUDIO GENÉTICO
El defecto primario en esta patología es una mutación inactivante en el gen SLC12A3 localizado en el cromosoma 16, que codifica el transportador Na-Cl sensible a tiazidas del túbulo contorneado distal27-32. La mutación intrón 9 +1G>T es característica de la etnia gitana. En nuestro caso, el estudio se realiza a la paciente y a sus hermanos.
Caso clínico
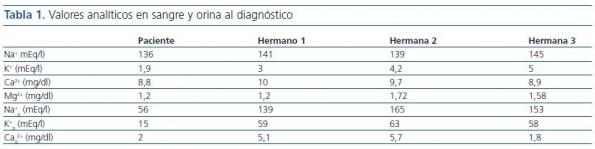
Presentamos el caso de una niña gitana de 8 años que estando previamente sana fue remitida a la consulta por dolor abdominal continuo y difuso de pocas horas de evolución acompañado de vómitos, febrícula y disuria. En la tabla 1 se muestran los valores analíticos en sangre y orina al diagnóstico.
En el estudio genético de la paciente se detectó que era homocigota para la mutación intrón 9 +1G>T en el gen SLC12A333-39.
Se realizó un estudio genético familiar con los siguientes resultados:
- Hermano 1: homocigoto para la mutación.
- Hermana 2: homocigoto normal, no portadora.
- Hermana 3: heterocigoto, portadora de la mutación.
DIAGNÓSTICO DE HIPOMAGNESEMIA
La forma más simple de evaluar la deficiencia de magnesio es con la medición de la concentración plasmática de magnesio40-43. Con respecto a esto, hay dos consideraciones importantes. El 30% del magnesio se liga a la albúmina; por tanto, la hipoalbuminemia puede producir una «seudohipomagnesemia ». Por otro lado, la mayor cantidad de magnesio en el organismo se localiza en el compartimiento intracelular. Por tanto, una persona puede tener un valor de magnesio plasmático normal y, aun así, presentar una deficiencia de magnesio intracelular y tener signos de hipomagnesemia44-47; esto se conoce como deficiencia funcional de magnesio. El magnesio ionizado libre en el plasma es la forma fisiológicamente activa y, además, la forma que mejor refleja las reservas intracelulares de magnesio. Por desgracia, en la práctica clínica actual no se cuenta con un examen de laboratorio que pueda medir las concentraciones plasmáticas de magnesio libre.
Una forma de evaluar la deficiencia funcional de magnesio en pacientes con concentraciones de magnesio plasmático normales, pero en los que se sospecha una deficiencia de magnesio, es por medio de la medición del magnesio plasmático después de una carga de magnesio. En primer lugar, se mide la excreción basal de magnesio en orina de 24 h. Después, se administra una infusión de 7,5 g de sulfato de magnesio en 8 horas y posteriormente se mide la excreción de magnesio en 24 h. Si el paciente excreta <70% de la carga de magnesio más la excreción de magnesio basal, esto se considera como deficiencia funcional de magnesio.
Si la causa no es aparente, la distinción entre pérdida renal y pérdida gastrointestinal de magnesio se puede hacer midiendo la cantidad de magnesio en una muestra de orina de 24 h o calculando la fracción de excreción de magnesio en una muestra de orina obtenida al azar. Una EFMg mayor del 3% o más de 1 mmol (24 mg) de magnesio en orina de 24 h indicarían pérdida renal de magnesio.
TRATAMIENTO DE LA HIPOMAGNESEMIA
En general, los pacientes con hipomagnesemia deben seguir una dieta rica en magnesio y la causa de hipomagnesemia debe ser tratada, de ser posible48.
Si el paciente es asintomático o la hipomagnesemia no es grave (magnesio plasmático >1 mg/dl), la vía oral es la ruta de elección, preferiblemente con preparaciones de liberación prolongada, como el cloruro de magnesio o el lactato de magnesio. En casos sintomáticos o cuando la concentración de magnesio es <1 mg/dl, la ruta intravenosa es la preferida. La preparación de elección es el sulfato de magnesio.
Se deben monitorizar los valores de magnesio plasmático buscando signos de toxicidad, como oliguria, depresión de conciencia y arreflexia. Los pacientes con insuficiencia renal deben recibir el 50% de la dosis si la creatinina sérica es mayor de 2. En casos de toxicidad, el antídoto es cloruro de calcio o gluconato de calcio intravenoso.
Los pacientes con hipomagnesemia inducida por diuréticos que por alguna razón no puedan interrumpirlos pueden beneficiarse del uso de amilorida, que puede disminuir la excreción de magnesio en el túbulo distal. Al parecer, amilorida causaría hiperpolarización de la membrana celular, lo cual favorecería la producción del potencial transmembrana necesario para la reabsorción de magnesio.

Tabla 1.



