






The adaptive immune response forms the basis of allograft rejection. Its weapons are direct cellular cytotoxicity, identified from the beginning of organ transplantation, and/or antibodies, limited to hyperacute rejection by preformed antibodies and not as an allogenic response. This resulted in allogenic response being thought for decades to have just a cellular origin. But the experimental studies by Gorer demonstrating tissue damage in allografts due to antibodies secreted by B lymphocytes activated against polymorphic molecules were disregarded.
The special coexistence of binding and unbinding between antibodies and antigens of the endothelial cell membranes has been the cause of the delay in demonstrating the humoral allogenic response. The endothelium, the target tissue of antibodies, has a high turnover, and antigen–antibody binding is non-covalent. If endothelial cells are attacked by the humoral response, immunoglobulins are rapidly removed from their surface by shedding and/or internalization, as well as degrading the components of the complement system by the action of MCP, DAF and CD59. Thus, the presence of complement proteins in the membrane of endothelial cells is transient. In fact, the acute form of antibody-mediated rejection was not demonstrated until C4d complement fragment deposition was identified, which is the only component that binds covalently to endothelial cells.
This review examines the relationship between humoral immune response and the types of acute and chronic histological lesion shown on biopsy of the transplanted organ.
La respuesta inmune adaptativa constituye la base del rechazo del aloinjerto. Sus armas lesivas son la citotoxicidad celular directa o los anticuerpos. La primera, identificada desde los inicios del trasplante de órganos y la segunda, limitada al rechazo hiperagudo por anticuerpos preformados y no como respuesta alogénica. Ello permitió mantener durante décadas que la respuesta alogénica tenía solo un origen celular. Pero se ignoraron los trabajos experimentales de Gorer que demostraban daño tisular en aloinjertos por anticuerpos secretados por linfocitos B activados frente a moléculas polimórficas.
La especial convivencia de unión y desunión entre anticuerpos y antígenos de membrana de células endoteliales ha sido la causa que retrasó la demostración de la respuesta alogénica humoral. El endotelio, que es el tejido diana de los anticuerpos, tiene un turnover alto y la unión antígeno-anticuerpo no es covalente. Si las células endoteliales sufren el ataque de la respuesta humoral, eliminan rápidamente de su superficie las inmunoglobulinas mediante shedding o internalización y, a la vez, degradan los componentes del complemento por la acción de MCP, DAF y CD59. Así, la presencia de las proteínas del complemento en la membrana de las células endoteliales es pasajera. De hecho, la forma aguda de rechazo por anticuerpos no se demostró hasta identificar el depósito del fragmento C4d del complemento, que es el único de unión covalente a las células endoteliales.
Esta revisión analiza la relación entre la respuesta inmune humoral y los tipos de lesión histológica aguda y crónica de la biopsia del órgano trasplantado.
Renal biopsy is the gold standard diagnostic test for acute rejection (AR) after renal transplantation (RT). Distinctively, it shows an infiltration of mononuclear cells (T lymphocytes), considered specific by the Banff classification when it affects the tubules (tubulitis) and/or the endothelium (endothelitis).1,2 Based on this, during the first four decades of RT, the cell theory remained the sole theory for AR, and humoral response was limited to hyperacute rejection, caused by preformed antibodies against HLA class I antigens and not secondary to the response of the recipient.3,4
However, during this long time the experimental work of Gorer was unknown. His studies demonstrated the formation of antibodies against H-2 histocompatibility antigens in 21 of 22 mice, after implantation of allogeneic sarcoma cells and in skin allografts, in response to antigen stimulation.5–7 Additionally, Morris showed the presence of cytotoxic antibodies following RT in man.8
Not until the early 1990s did Feucht show the presence of C4d deposits in peritubular capillaries as a mark of complement system activation by the action of anti-HLA antibodies.9,10 Subsequently, the work of Terasaki11,12 and the successive contributions of the Banff classification13,14 cleared the way for the diagnosis of humoral AR. Although doubts remain about the cell and molecular pathways regulating antibody-mediated rejection, current understanding of their immunobiology shows that activation of B lymphocytes induced by polymorphic molecules (HLA or non-HLA) results in the formation and secretion of donor-specific antibodies (DSA) that damage the allograft.15,16 This review examines the relationship between the immune response and the histology of humoral rejection in RT.
Immune systemThe foundations of what is now known as the humoral immune response were established millions of years ago when the first living beings shared their habitat with pathogens that posed a threat to their survival. This evolutionary challenge led to the creation of a defense infrastructure known as the immune system (IS).17
The most elementary invertebrates developed an IS similar to phagocytosis and the more complex invertebrates developed an IS composed of molecules (cytokines, complement system and acute phase proteins) and eminently phagocytic cells (neutrophils, monocytes, macrophages, eosinophils, basophils, mast cells, natural killer [NK] and dendritic cells). These lacked immunological memory, had limited progeny, a relatively long life and high efficiency receptors encoded in the germline, which only recognized microorganism structures called “pathogen-associated molecular patterns”; they were therefore unable to recognize other molecular differences. This response is known as innate immunity.18
Evolutionary pressure, in a continuous process of “adapt or die”, allowed vertebrates to complete their IS, adding a new identification and defense infrastructure known as adaptive or acquired immune response,18,19 consisting of T and B lymphocytes. The distinctive element of this system is its ability to generate membrane receptors by random gene rearrangements; representing a high capacity to form very many different receptors that can recognize a wide variety of antigens. These cells are also equipped with immunological memory and the ability to proliferate, on encountering a recognized antigen, and form a clone with a specific receptor. Thus, their repertoire in the total population is so broad that there is an increased probability that an antigen will present to a particular lymphocyte, bind to its receptor and induce activation and proliferation. This process is known as clonal selection and is a basic feature of the adaptive immune response for recognition of molecular differences; in our case, provided by the allograft.
Given the large number of cells and molecules forming the IS, its ability to recognize and respond is very varied and is carried out by neutralization, opsonization, phagocytosis and humoral and cell lysis. The IS uses the most effective mechanism for each threat.
Which of these mechanisms comprise the most effective response against an allograft? Neutralization and cellular and humoral lysis; precisely, the mechanisms that the IS launches against viruses.
Humoral responseBrain death and ischemia induce an inflammatory response characterized by an infiltration of polymorphonuclear cells and macrophages, with increased expression of selectins and complement factors C3a and C5a.20 The cells injured by ischemia release materials (damage-associated molecular patterns – DAMPS), which are ligands of Toll receptors. Epithelial, endothelial and mesenchymal cells have Toll receptors that, stimulated by their ligands, are the starting point of biochemical signals that activate the innate immune response.21 Damaged endothelial cells alter their permeability and anticoagulant function, and the altered molecules induce an inflammatory response and complement activation (in this case via the lectin pathway).22
DAMP molecules stimulate the dendritic cells that process and express donor HLA antigens in their molecules. In conclusion, the allograft carries a load of activity of the innate immune response that intensifies after implantation through ischemia-reperfusion,23,24 a process that is the precursor to the adaptive immune response of the recipient, which provokes allograft rejection.
The allogeneic humoral response is initiated by allograft-antigen recognition by the activated B cell that proliferates to form an antigen-specific clone, which actively secretes antibodies and generates memory cells.25
Several B cell activation pathways exist (independent of Th lymphocytes and vs disaccharides), but it is the Th cell-dependent pathway that is associated with allogeneic transplant. This pathway characterizes responses to proteins such as HLA. These responses require ligation of the antigen receptor plus the delivery of T-cell help, particularly through CD40/CD40 ligand interactions.25
Since B cell activation leads to the formation and secretion of antibodies as a harmful weapon, its detection in blood is used to assess humoral response. But, surprisingly, although the detection technique has been perfected,26 the presence of antibodies against HLA antigens is low in the first phase of transplantation, despite immunosuppressive therapy that is more effective against T cells than B cells. Some have interpreted this as a reflection of the lack of a B-cell response to allogeneic stimulation,27 and others as the possibility that the response is higher and that antibodies are absorbed and cleared by the allograft.28
A more direct procedure for assessing allogeneic humoral response by calculating the frequency of antibody-secreting B cells using the ELISPOT technique has been proposed. A trial of this method in recipients of allogeneic RT, using as a target fibroblasts cultured from the donor instead of purified antigens, showed that although none of the nine patients in the study had DSA in serum before or after transplantation, all exhibited an increased number of DSA-secreting cells directed against donor HLA class I antigens eight weeks post-transplant. This supports the concept that the allogeneic humoral response is more frequent than what is deduced by the detection of antibodies in blood.29
Infrastructure of the humoral responseThe Banff classification bases diagnosis of humoral AR on the presence of serum DSA, on histological criteria of acute tissue damage of the vascular wall (intimal or transmural arteritis and acute thrombotic microangiopathy) and on the interaction between antibodies and the vascular endothelium (C4d deposition in peritubular capillaries, microvascular inflammation and increases in endothelial activity and endothelial activation and transcripts (ENDATs).30,31
The specific infrastructure of the immune response causing these histological lesions consists of B lymphocytes, antibodies (anti-HLA or others), inflammatory cells (neutrophils, monocytes-macrophages and NK) and the complement system and, as a target, the endothelial cells.
- 1.
B lymphocyte. This is the cell that generates antibodies. Two types of B lymphocyte antibody generators exist.32,33 B1 resides in the pleura and peritoneum and produces low affinity antibodies irrespective of the Th lymphocytes; and B2 permanently circulates through the secondary lymphoid organs to find an antigen that activates and expands it (clonal proliferation). Once activated it interacts with the Th lymphocyte receptor to present the antigen in HLA class II molecules to it and activate it. In addition, the B cell produces cytokines that stimulate the T cell. Thus, the B lymphocyte plays a prominent role in the activation and development of T memory cells.34
The activated B lymphocytes form extrafollicular plasmablasts that produce low affinity antibodies or migrate to the germinal center where somatic hypermutation of the variable-region immunoglobulin genes, immunoglobulin class switching and plasma and memory cell generation manufacturing and secreting IgG antibodies originate. For the germinal centers of the B cells to form, the presence of follicular T lymphocytes is necessary.35
Notably, in animal36 and human37 models of transplantation the formation of tertiary lymphoid organs in the allograft has been seen, suggesting that B cells can be activated directly in the transplanted organ.
A subpopulation of B cells inhibiting the immune response has also been demonstrated.38 This regulatory function is done by IL-10 secretion.
- 2.
Antibodies. These are glycoproteins with a heavy and light double chain symmetrical structure, part of the defense system against pathogens. In allogeneic RT they are generated in response to antigenic stimuli caused by polymorphic molecular differences and have been described against HLA, MICA, ABO, vimentin, phospholipids, stress proteins and the angiotensin II AT1 receptor in relation to AR and chronic rejection.39 The most frequently generated antibodies are against HLA molecules, because they are the most polymorphic, and being expressed on the endothelial cell membrane makes them very vulnerable.
Understanding the role of antibodies in RT requires focusing its analysis on the following:
- a.
Immunoglobulin chains are joined together covalently. This type of binding is formed by non-metallic atoms with many electrons in their periphery and a tendency to attract even more; each atom is attached to another by exchanging an electron to form a very strong bond.
- b.
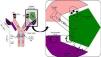
Antigen binding to the antibody is noncovalent, but is regulated by electrostatic forces, hydrogen bonding and Van der Waals and hydrophobic forces forming a reversible bond; temperature sensitive, antigen/antibody proportional, pH and ionic strength of the medium (Fig. 1).
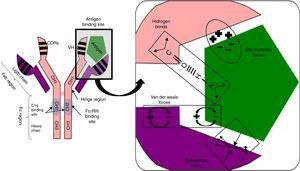 Fig. 1.
Fig. 1.Non-covalent antigen–antibody binding regulated by hydrogen bonds and electrostatic, van der Waals and hydrophobic forces. Structure of an IgG antibody. The antigen binding sites are formed by the juxtaposition of variable light chain (VL) and heavy chain (VH) domains. The CH2 domain of the Fc region is the binding site for C1q or FcγR of inflammatory cells. CDRs: complementarity domain regions. HC: heavy chain.
- c.
The hinge region, located between the CH1 and CH2 domains, provides flexibility to the immunoglobulin to guide each of its arms to the antigen binding.
- d.
Fab region, comprising two antigen binding arms, each formed in both the heavy and the light chain by one variable and one constant domain. The variable is so called because in each chain it has three segments of variability formed by ten amino acid residues that differentiate antibodies produced by one particular B cell clone from another (Fig. 1).
The three variable segments of the heavy and light chains combine to form a three-dimensional antigen binding surface space. Since this surface is complementary to the antigen binding region (like a key and lock), they are called complementarity determining regions (CDRs) or CDR1, 2 and 3. Those with the greatest variability and antigen contact are CDR3.
- e.
Fc region, consisting of two or three heavy chain constant domains (Fig. 1), according to the immunoglobulin serotype (three for IgM and two for the remainder) and mediating the effector functions of the antibody on binding to the C1q complement fraction and to cells with Fc region receptors (FcRs) having a polypeptide α chain with a polymorphic character that determines binding to the Fc region.
- a.
- 3.
Inflammatory cells. The pathophysiology of humoral AR begins with the binding of DSA to HLA and non-HLA allograft antigens, expressed on the endothelial cell membrane. This process generates two-way attraction and activation of inflammatory cells (neutrophils, macrophages and NK cells):
- a.
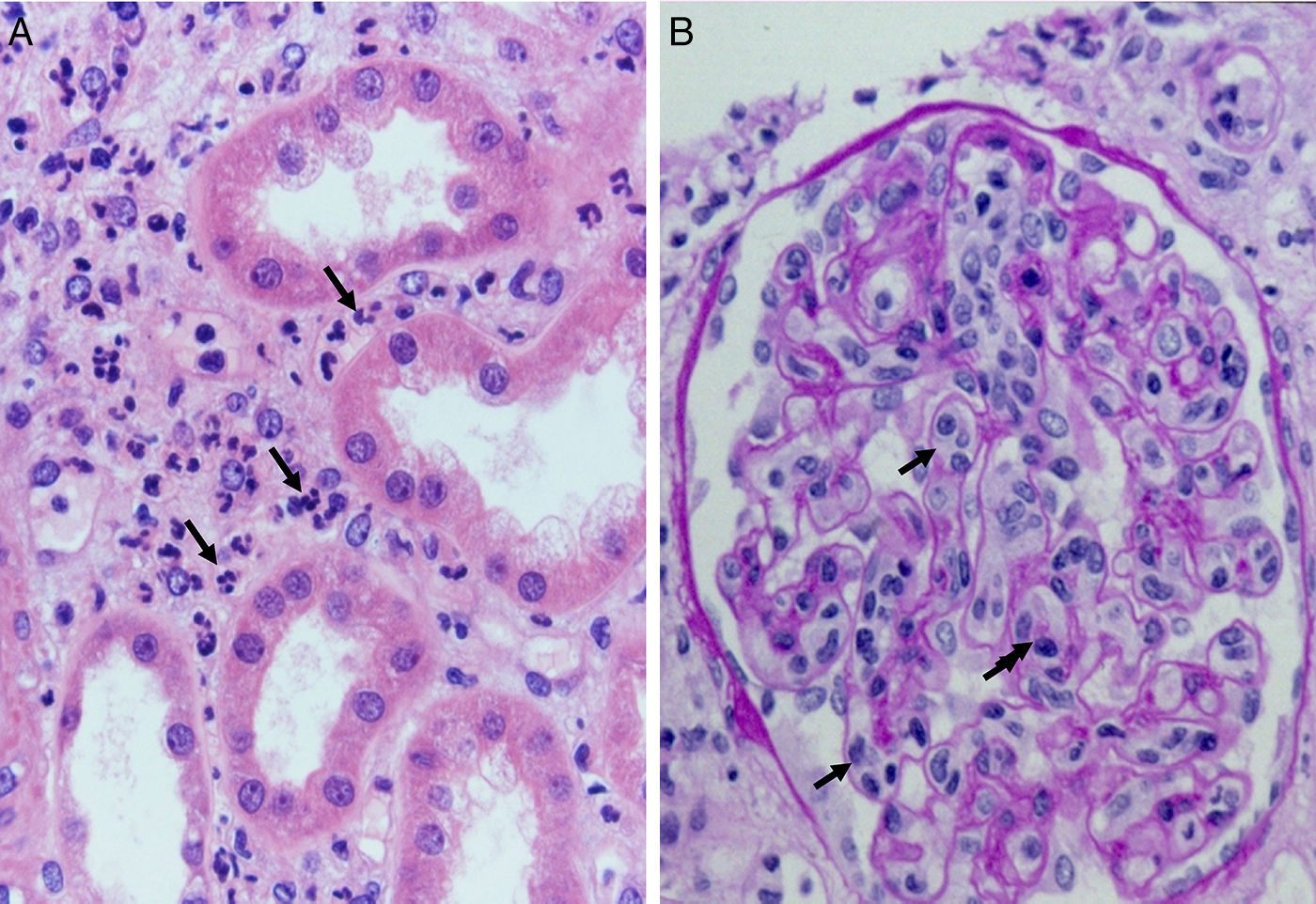
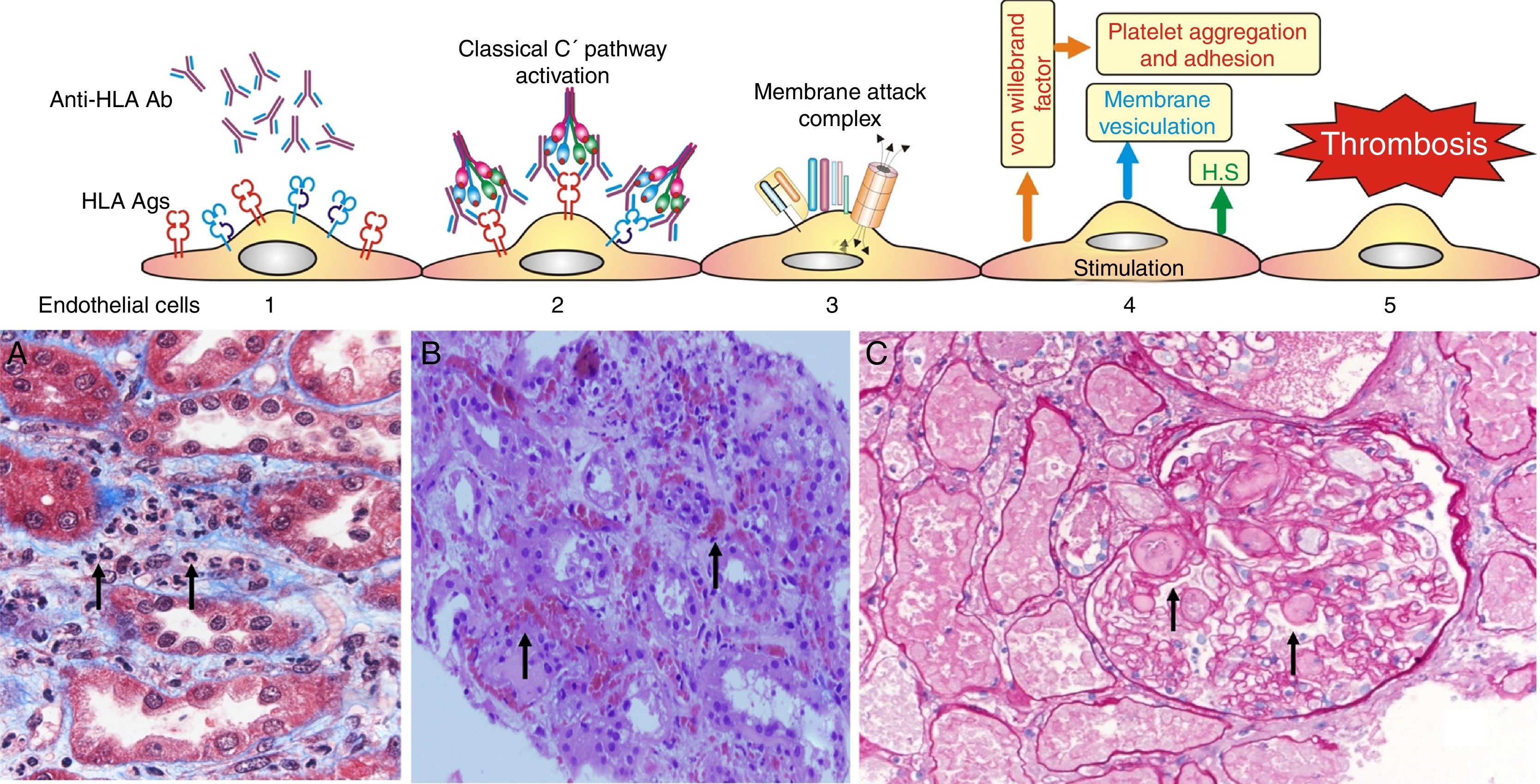
Complement dependent. Complement activation by cytotoxic antibodies bound to the HLA antigen of the endothelial cell membrane generates the C3a and C5a fractions, which are potent opsonins. By chemotactic gradient, they attract inflammatory cells to the peritubular (capillaritis) (Fig. 2A) and glomerular capillaries (glomerulitis) (Fig. 2B) and activate them through interaction with their cognate receptors (C3aR and C5aR) to secrete enzymes and pro-inflammatory cytokines.
- b.
Complement independent. IgG isotype antibodies act as ligands for endothelial cell membrane molecules of the glomerular and peritubular capillaries and are bound by the CH2 domain of the Fc region to inflammatory cells having FcγRIII that induce antibody-mediated cell lysis.
The role of NK cells in antibody-mediated cytotoxicity should be emphasized. Once bound to the CH2 domain of the Fc region by their FcγRIII, they are activated and secrete INF-γ and the content of their granules with which they cause cell lysis.
- a.
- 4.
Complement system. Activation of the classical complement pathway following an episode of humoral response in allograft organ transplantation is initiated by the binding of the first fragment, C1q, to the CH2 domain of the Fc region of the IgG isotype immunoglobulins. Not all IgG subclasses activate it; only IgG3 and IgG1 (in order of intensity), IgG2 does so weakly and IgG4 has no reactivity.40 This difference is determined by the CH2 domain polymorphism.
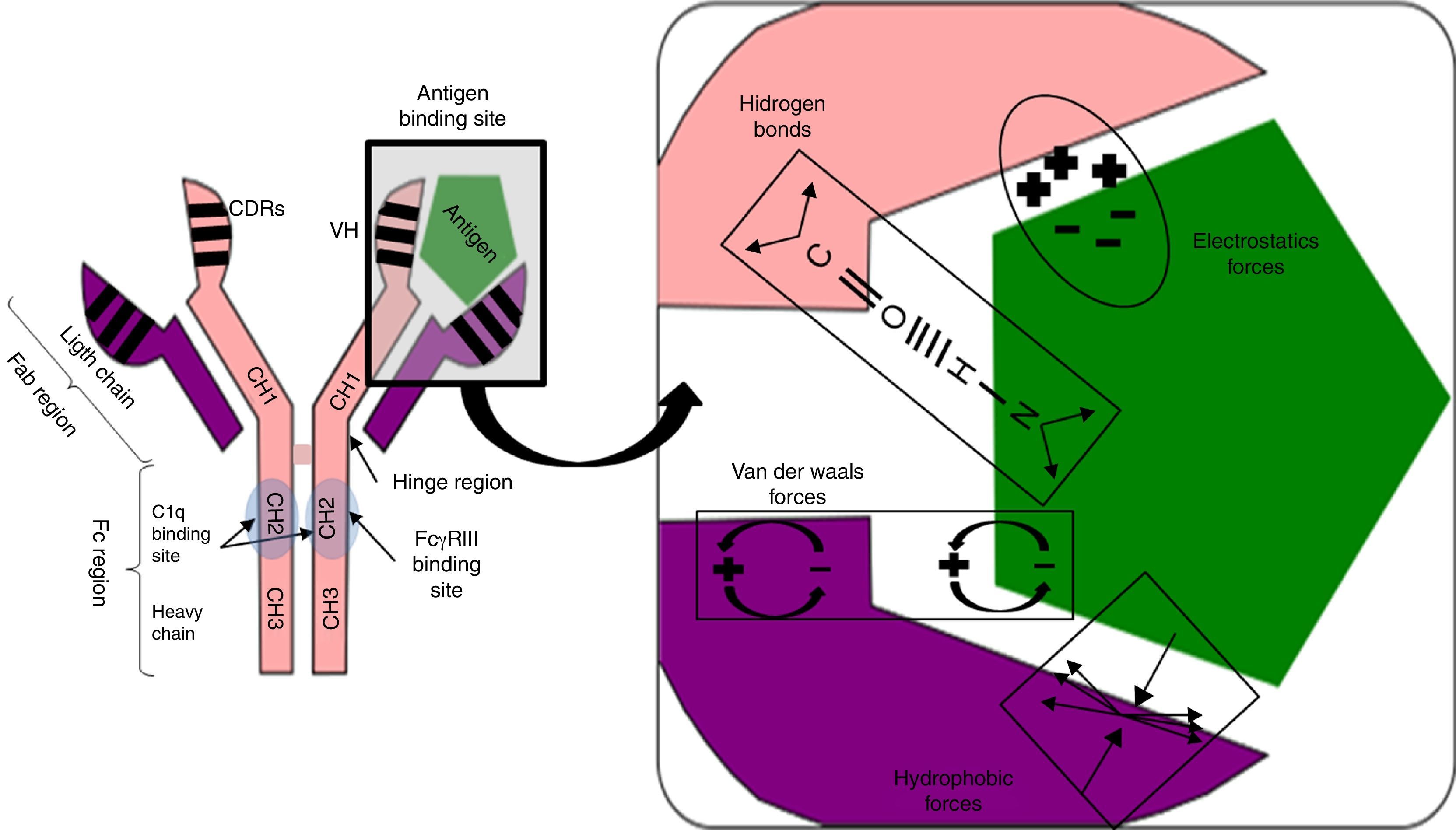
Based on protein engineering studies of mouse IgG2b, three charged amino acids (glycine 318, lysine 320 and lysine 322) located on one β strand of CH2 were proposed as constituting the essential C1q binding motif.41 But the binding is not sufficient to activate complement, because this motif is present in all IgG subclasses. It needs something else.
Studies with mutants of IgG subclasses have shown that the presence of lysine at position 276, very close to the binding motif of C1q (glycine 318, lysine 320 and lysine 322) gives the IgG3 its ability to initiate complement activation and in IgG1 the presence of proline at position 291. In contrast, IgG4 is unable to activate complement, despite binding to C1q, due to the presence of a serine residue at position 331. This confirms that C1q binding to the CH2 domain of the Fc region of the immunoglobulin only is not sufficient to activate complement, but rather the presence of certain amino acids at various positions in the CH2 domain of the Fc region determines whether or not the IgG subtype activates it.41,42
IgG antibodies have a longer half-life than other proteins because the neonatal Fc receptor binds to the IgG after being endocytosed by the cell and instead of being degraded is recycled to the cell surface.43 Mice with a deficit of neonatal Fc receptor have decreased circulating IgG levels and a reduced immunoglobulin half-life. Immunoglobulin administration to treat humoral rejection has among other properties that of binding to neonatal receptors and saturating them, thereby inhibiting the interaction of endogenous IgG antibodies with the neonatal Fc receptor, favoring their disappearance.44
In addition to the chemotactic function cited, complement has the following functions:
- a.
C3b, iC3b and C3d opsonins promote cell lysis.
- b.
The membrane attack complex (C5b-9) lyses cells or opsonized pathogens.
- c.
Experimental models have shown that uncontrolled complement activation increases the T-cell reactivity through costimulatory signals on antigen-presenting cells and T lymphocyte allograft-antigen recognition.45,46
- d.
The C3a and C5a fractions stimulate differentiation of Th0 into Th1 cells; and C3aR and C5aR signaling inhibits development of regulatory T cells.47
- e.
Allograft cells that are opsonized by complement fractions have greater interaction with T cells, suggesting that complement-mediated cell adhesion may be important in tissue damage mediated by T cells.48
The onset of formation of antibodies against allograft antigens is complement-dependent.49,50
Antibody target cellEndothelial cells are the antibody targets in the humoral response. Thus, humoral rejection is a model of endothelial dysfunction.
Endothelial cells form a functional unit with the underlying smooth muscle cells and interstitial matrix. They control passage of solutes, macromolecules and blood cells to tissues. This process is regulated by molecules which increase (histamine, thrombin, TNF-α, bradykinin, etc.) or lower (heparan sulfate, prostaglandins, catecholamines, natiuretic peptide, β-adrenergic receptor stimulators, etc.) vascular permeability.
They also contribute to the hemostatic balance by separating plasma coagulation factors from coagulation activators present in the interstitial matrix secreted by smooth muscle cells. If the barrier effect is damaged, they are exposed to each other, initiating a process of activation of plasma factors IX and X by the VIIa tissue complex; and in response to thrombin, platelets express receptors for the von Willebrand factor and platelet aggregation is activated. In this way, damage to the vessel wall eventually causes thrombotic microangiopathy. The kidney has a large area of endothelium in the peritubular capillaries and glomeruli, which will suffer the most significant damage by the action of antibodies. The induction pathways by which antibodies cause injury to endothelial and smooth muscle cells are still being studied, although more is known about those caused by class I than by class II antibodies.
Why have there always been difficulties diagnosing acute humoral rejection?Endothelial cells express HLA class I antigens natively on their membranes and after stimulation with INF-γ class II; and the kidney has a large endothelial surface on its peritubular and glomerular capillaries upon which the anti-HLA antibodies may act.
The question in autoimmune glomerular diseases is, why were there no serious difficulties proving their autoimmune origin by biopsy, but why are there difficulties diagnosing humoral AR? The response requires that the pathophysiological differences and, especially, the target tissue and its turnover must be considered. In autoimmune glomerular diseases, in addition to their pathophysiological differences, immune complexes are located in low turnover tissues such as the subendothelium in lupus nephritis or in the basement membrane in Goodpasture syndrome and membranous glomerulonephritis. Immune complexes are retained for long periods in these low-turnover tissues, long enough to be revealed on renal biopsy.
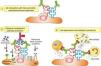
In humoral AR, however, antibodies are directed against HLA antigens of endothelial cells, which have a high turnover. If they are attacked by the humoral response they can quickly remove surface immunoglobulins by shedding and/or internalization51,52; and at the same time inhibit the activation of the complement system in the early stages of the process and in the late stages degrade their components by the action of membrane cofactor protein53 (MCP), decay accelerating factor54 (DAF) and CD5955 (Fig. 3). Therefore, the presence of complement proteins in the endothelial cell membrane is transient; and since the onset of immune damage precedes clinical signs of rejection, when the biopsy is performed the humoral origin of the attack is not detected.
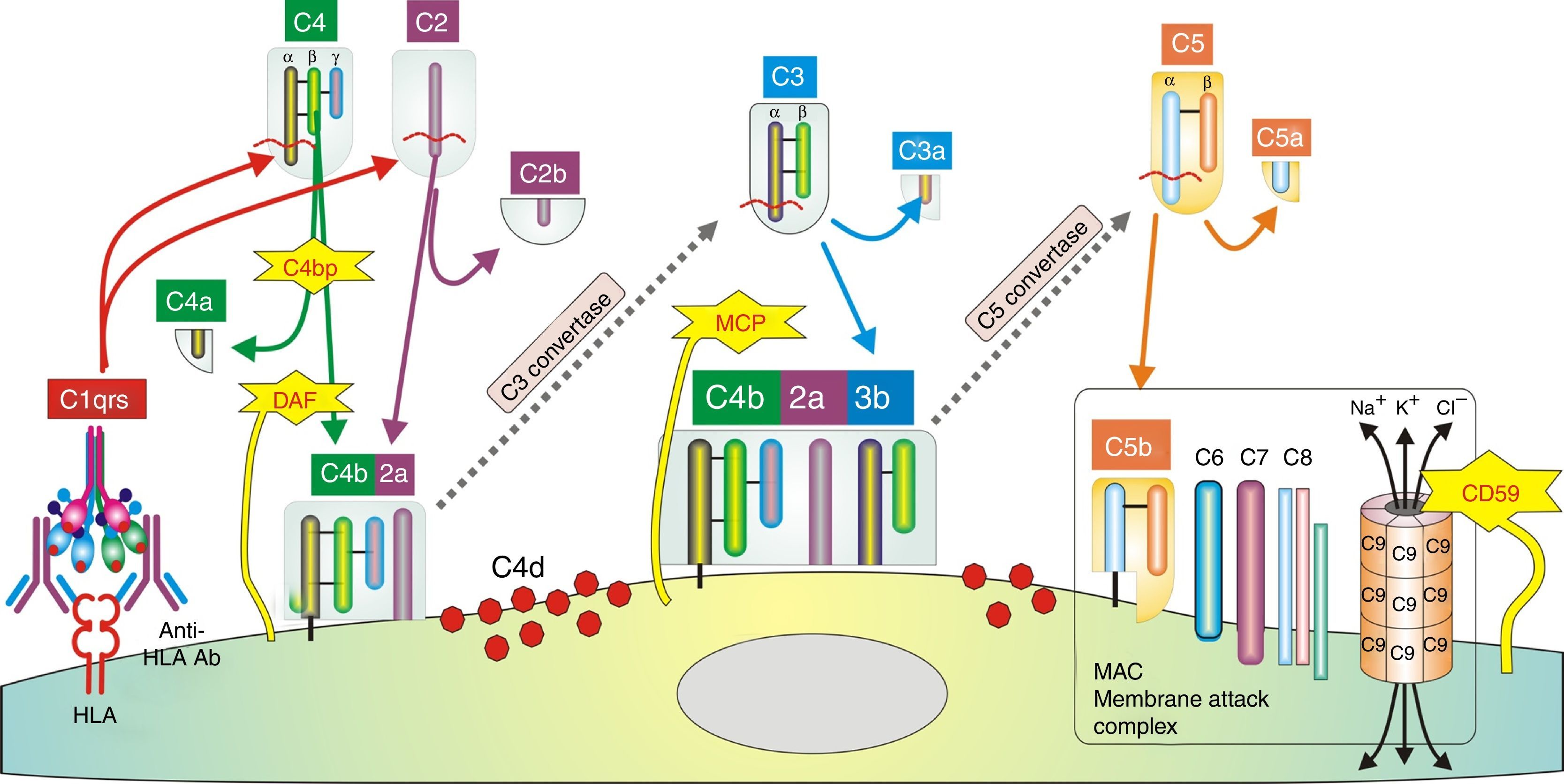
Inhibition of classical complement activation by membrane-bound and circulating control proteins. The serum protein C4bp neutralizes C4. DAF blocks the action of C4b, impeding digestion of C2 to C2a and C2b. MCP acts as cofactor I, degrading C4b to C4c and C4d. Finally, CD59 is anchored to the membrane and prevents polymerization of C9. DAF: decay accelerating factor. MCP: membrane cofactor protein.
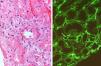
How did the C4d technique enable diagnosis of humoral AR? Because this complement fraction binds to endothelial cells covalently through a thio-ester bond to form a stable bond, resistant to shedding. This is quite the opposite of what happens with the C4c fragment, which degrades quickly. Therefore, C4d deposition could demonstrate the presence of a previously undetectable humoral response (Fig. 4). Nevertheless, the latest Banff classification considers as histological data of acute or chronic humoral AR evidence of endothelial damage by the interaction of antibodies on endothelial cells in patients with circulating DSA.13 It thus recognizes the evidence of humoral rejection with negative C4d.30 The reasoning has been if the antibodies act on endothelial cells, they can stimulate the expression of activation genes (ENDATs) that produce transcripts that can be determined by microarrays. In this way, a phenotype of humoral rejection could be identified with circulating antibodies with negative C4d. Thus, kidneys with a high expression of ENDATs in the graft and cytotoxic anti-HLA antibodies in blood showed histological lesions compatible with antibody-mediated rejection. The conclusions are: a high expression of ENDATs with circulating antibodies predicts graft loss with higher sensitivity (77 vs 31%) and lower specificity (71 vs 94%) than the presence of C4d. However, high ENDAT expression was not an indicator of graft damage or eventual graft loss in patients who lacked anti-HLA antibodies.56
Antibodies and histologic lesions in allogeneic transplant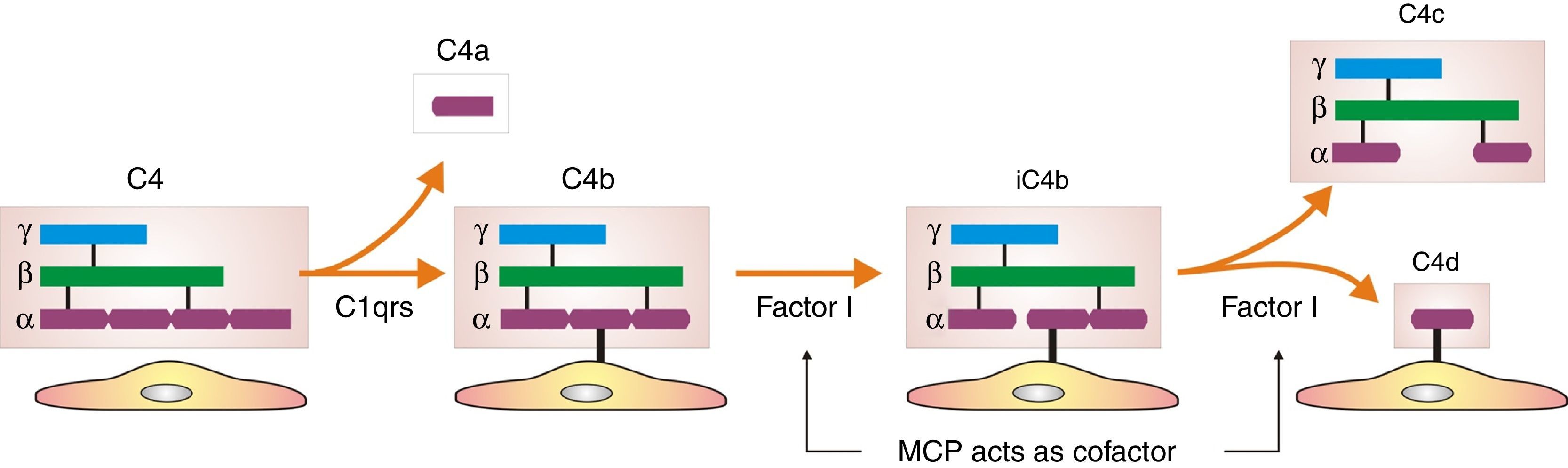
Cytotoxic antibodies in organ transplantation cause endothelial damage by Fig. 5:
- 1.
Activation of the complement system.
- 2.
Direct action.
- 3.
Recruitment of inflammatory cells via Fc receptors (antibody-mediated cell immunity).
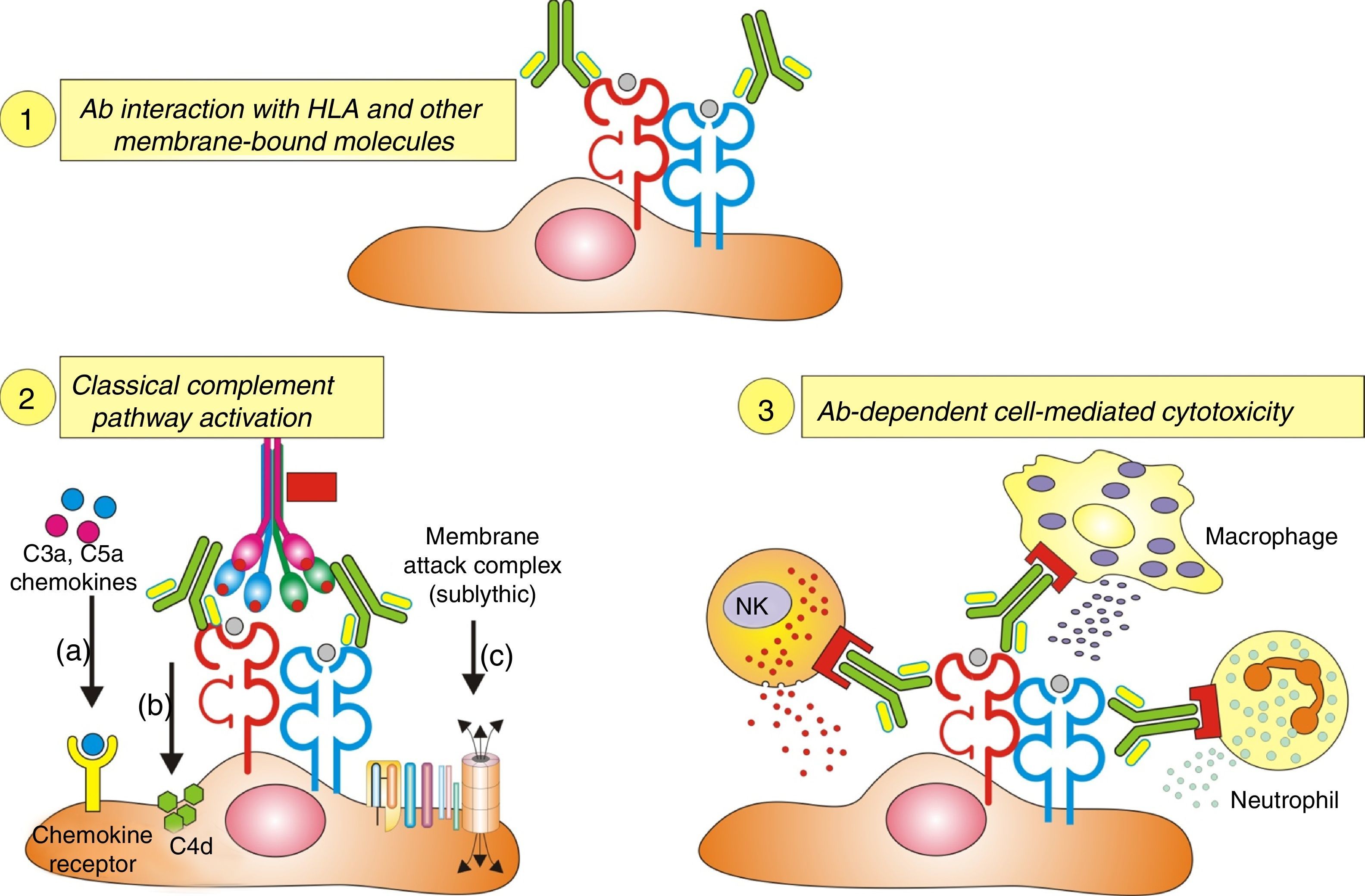
Ligation of HLA molecules by high titers of anti-HLA antibodies can generate: (1) Direct tissue damage by increasing the expression of fibroblast receptors (FGFR) and cell proliferation. (2) Activation of the classic complement pathway. (3) Cytotoxicity mediated by antibodies and Fc receptors causing capillaritis and/or glomerulitis. FGF: fibroblast growth factor.
- a.
Hyperacute rejection. This is the most genuine example of severe endothelial damage in the allogeneic transplant, induced by preformed antibodies that activate complement through the classical pathway.
Once the allograft vessels are unclamped and the blood starts flowing in the transplanted organ, the antibodies bind to HLA class I antigens expressed on the membrane of endothelial cells of glomeruli and microvessels. Complement is activated and the graft immediately takes on a limp texture and mottled color that is usually a result of irreversible damage.
Experimental data suggest the following pathophysiological sequences (Fig. 6):
- •
The endothelial cell membrane is coated with a layer of heparan sulfate. This proteoglycan maintains a local anticoagulant environment by activating antithrombin III, a potent inhibitor of thrombin formation. It also participates in the regulation of endothelial barrier impermeability to the passage of cells and molecules since it is part of the union between endothelial cells and their cytoskeleton; and also through its electrical charge it rejects the plasma coagulation factors from the endothelial surface.
- •
Experimental data have shown that the exposure of porcine endothelial cells to human xenoreactive natural antibodies causes progressive release of heparan sulfate from their surface mediated by enzymatic cleavage of the protein core and/or glycosaminoglycan chains. In contrast, the supernatant from endothelial cells exposed to human serum for 4h contained intact proteoglycan, possibly reflecting vesiculation or cell lysis as well as proteoglycan fragments. The cleavage and release of endothelial cell proteoglycans appeared to be triggered by the binding of natural antibodies to endothelial cells and activation of complement.57
- •
Loss of heparan sulfate is accompanied by alterations in the shape and the cytoskeleton of the endothelial cells that disrupt monolayer integrity and lead to formation of intercellular gaps allowing the passage of cells and molecules, causing edema and interstitial bleeding visible on biopsy58 (Fig. 6B).
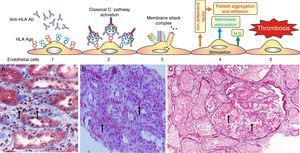 Fig. 6.
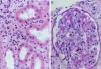
Fig. 6.Sequence of events in hyperacute rejection (explanation in the text). (A) Capillaritis (Masson trichrome). Presence of polynuclear leukocytes in the peritubular capillaries attracted by chemotaxis. (B) Interstitial hemorrhage (H&E). The release of heparan sulfate causes intercellular gaps via which the red cells reach the interstitium. (C) Thrombotic microangiopathy with cortical necrosis (PAS).
The loss of the endothelial barrier effect exposes the interstitial tissue VIIa complex and the plasma coagulation factors IX and X that are activated; and in response to thrombin, platelets express receptors for the von Willebrand factor and platelet aggregation is activated. This process results in thrombotic microangiopathy (Fig. 6C).
- •
The tubules receive oxygen and nutrients through the peritubular capillaries. Vascular injury causes necrosis of tubular epithelial cells and continuation of the process leads to tissue necrosis.
The presence of inflammatory cells, especially neutrophils, in the peritubular capillaries (capillaritis) and glomeruli (glomerulitis), is due to the chemotactic effect of the C3a and C5a complement factors that are very powerful opsonins attracting these cells to the site of injury, where they secrete pro-inflammatory cytokines.
- •
- b.
Acute humoral rejection. Promoted by a humoral memory response generated by prior exposure to HLA or other antigens, provided by the allograft and expressed on the endothelium of peritubular and glomerular capillaries.59–61
Rejection appears a few days after the transplant but if the patient receives induction with anti-lymphocyte antibodies its appearance is delayed by weeks.62 It is produced by the memory response to previous exposure to HLA antigens in the early phase of RT and in the late phase by noncompliance with immunosuppressive treatment63 (Fig. 7).
From the standpoint of prognosis, clear differences exist between humoral AR and hyperacute rejection. AR is reversible, while hyperacute rejection generally is not. However, the participants are the same (DSA and endothelial cells). Endothelial cells have the same characteristics in both cases. The difference is in the antibodies. In hyperacute rejection they are preformed, generally complement activators, of high concentration and with a high affinity for their alloantigen. Conversely, a decreased expression of these qualities will trigger a less severe rejection episode and we will be faced with AR. Obviously the scale is not so simple, but at present further explanation is not possible, since among other difficulties, neither the concentration nor the affinity of allospecific antibodies can be measured.
Direct complement-independent actionAntibodies cause direct tissue damage by acting as antigen agonists expressed on the endothelial cell membrane and induce pro-inflammatory and proliferative intracellular signals in both AR and chronic rejection.
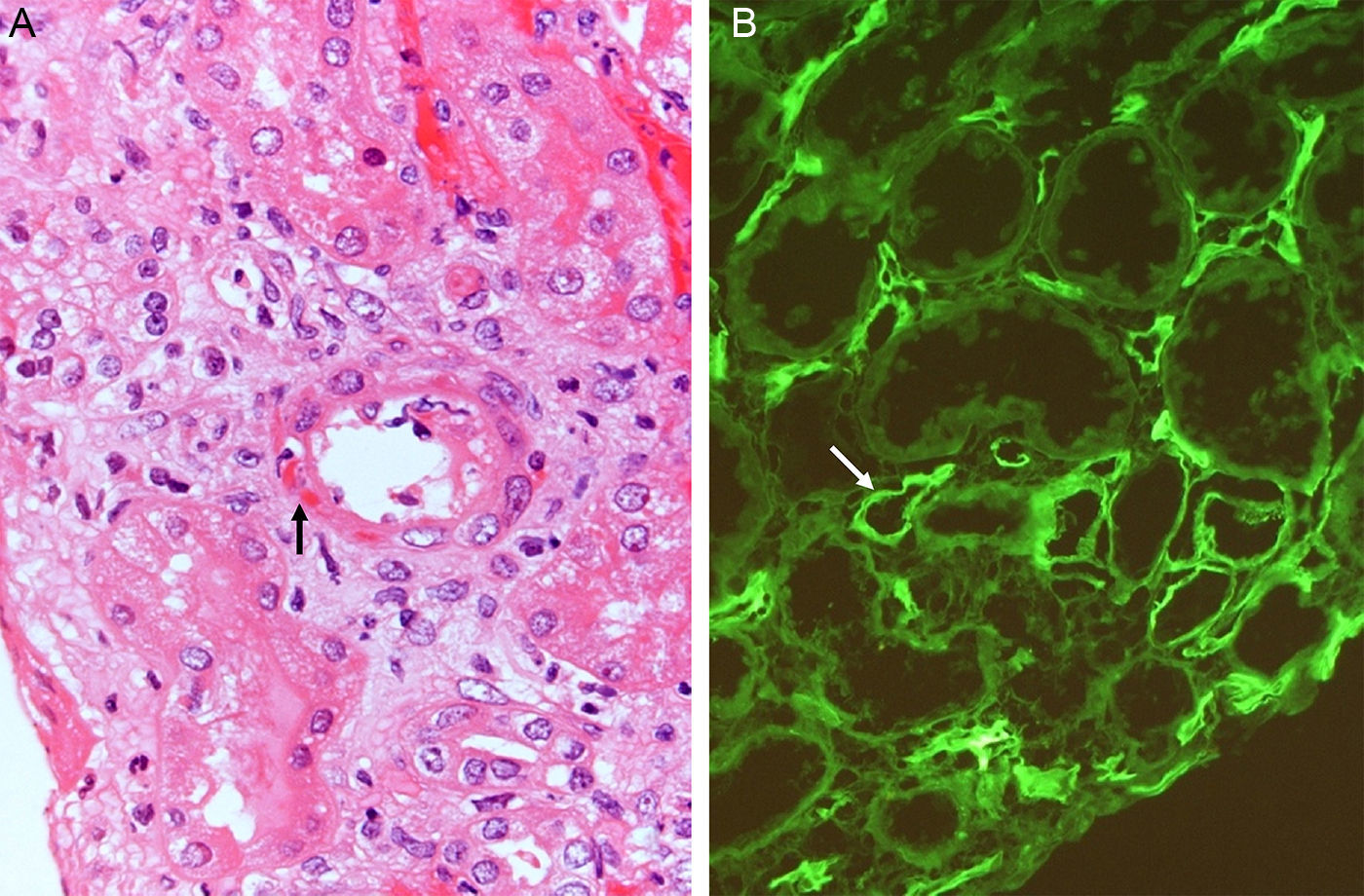
The most authentic injury resulting from this action is transplant vasculopathy (Fig. 8A). An obliterative chronic injury of the graft vessels caused by proliferation and hyperplasia of endothelial cells and smooth muscle which decreases the vessel size and causes ischemic damage and progressive worsening of renal function.
The pathophysiology can be summarized as: the anti-HLA class I antibodies bind as ligands to the HLA antigens expressed on the endothelial cells to induce stimulation of the mTOR pathway and S6 kinase phosphorylation (S6K) and S6 ribosomal protein (S6RP), promoting protein synthesis and cell proliferation.64,65 These data were reproduced in an experimental model of murine MHC-incompatible heart transplantation with continued administration of class I antibodies. The amount of antibody supplied correlates with increased phosphorylation of S6K and S6RP on the endothelial cells of the graft capillaries. A situation similar to that which occurred in human heart transplantation with antibody-mediated rejection in which the S6RP phosphorylation in endocardial biopsies is associated with the presence of circulating antibodies66 (Fig. 9). Additionally, antibodies that act as agonists of HLA class I antigens induce the expression of growth factor receptors of fibroblasts with an intensity dependent on the level reached in blood. In this case, the induced cellular pathway is the MEK-ERK signal that stimulates endothelial cell proliferation67 (Fig. 9).
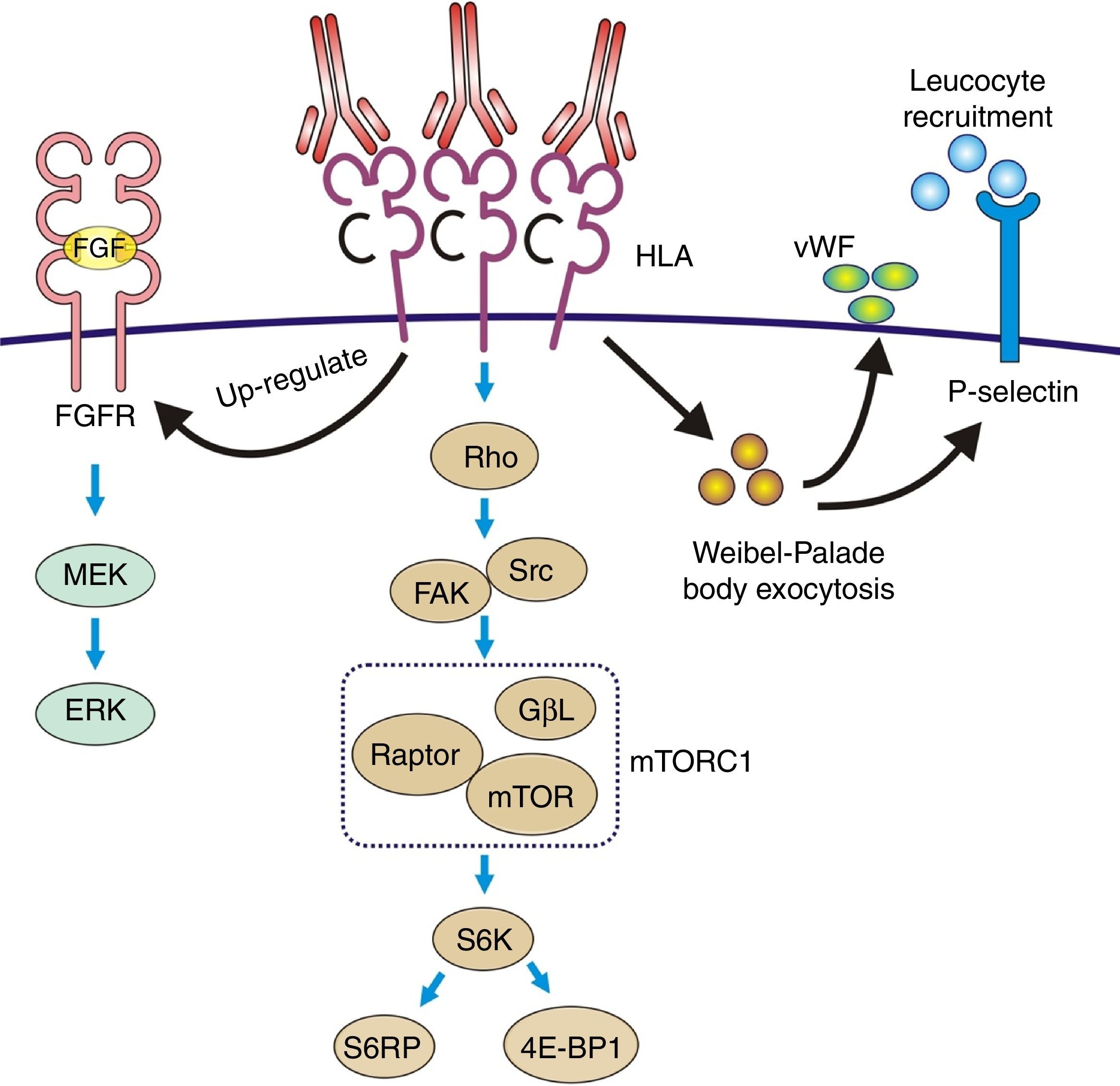
Pathophysiology of transplant vasculopathy. Antigen–antibody binding generates: (1) Increased expression of fibroblast receptors, which activate the MEK/ERK pathway and AP-1 and NF-kB, inducing cell proliferation. (2) The Weibel–Palade bodies secrete their contents of von Willebrand factor and P-selectin, which favors leukocyte recruitment. (3) Stimulation of the Rho pathway, which induces protein synthesis and cell proliferation.
The effect of class II antibodies is less known, although data suggest cell proliferation occurs through the S6 and S6RP activation pathway.64
Recent studies suggest NK cells play an important role in the regulation of allograft acceptance or rejection.68 Their role is not limited to that described thus far of killing and cytokine production, but they may be important in the development of transplant vasculopathy. In a model in which DSA were infused into immunodeficient Rag−/− mice that were grafted with heart allografts to which the DSA were directed, graft vasculopathy has been reproduced within four weeks. The mechanism of action of NK cells in the development of cardiac allograft vasculopathy is that they bind by their FcRs to the antibody Fc domain and are activated, secreting pro-inflammatory cytokines that induce proliferation of endothelial cells and smooth muscle.69,70 The study conclusions are:
- a.
NK cells are absolutely needed for the development of full-fledged vascular lesions in the grafts, as neither NK-depleted mice nor recipient mice genetically deficient for NK cells (Rag−/−γc−/− mice) developed transplant vasculopathy.
- b.
A role for the Fc portion of DSA is indicated in this model. This is because infusion of the F(ab′)2 fragment of DSA failed to induce vasculopathy in the graft.
- c.
Complement is dispensable. Transplant vasculopathy can be induced with noncomplement-fixing DSA or in C3-deficient Rag−/− mice in which complement activation is inhibited.
Others suggest that the assessment of the NK cell immunophenotype may contribute to define signatures of alloreactive humoral responses in renal allograft recipients.71
Recruitment of inflammatory cells via Fc receptors (antibody-mediated cell immunity)Antibody-activated endothelial cells express VCAM-1 and ICAM-1 adhesion molecules, which promote the adhesion of inflammatory cells72 that activate exocytosis of granules containing prothrombotic mediators, such as von Willebrand factor and P-selectin, by triggering calcium-mediated Weibel–Palade body exocytosis (Fig. 9). The biologically active complement split-product C5a adds a slight but significant increase to antibody induction of exocytosis. Crosslinking of HLA appears critical to stimulate exocytosis, because only the bivalent F(ab′)2 of one class I antibody W6/32 is effective in trigging exocytosis. Ligation of MHC class I molecules by antibodies also leads to a dose-dependent increase in the production of monocyte chemoattractant protein-1 and neutrophil chemoattractant growth-related oncogene α that attract macrophages to the graft.73
Transplant glomerulopathyThis is a histologically defined entity, associated with molecular pathways of DSA induction that are not well known.74 Along with transplant vasculopathy it is the most representative entity of chronic rejection.
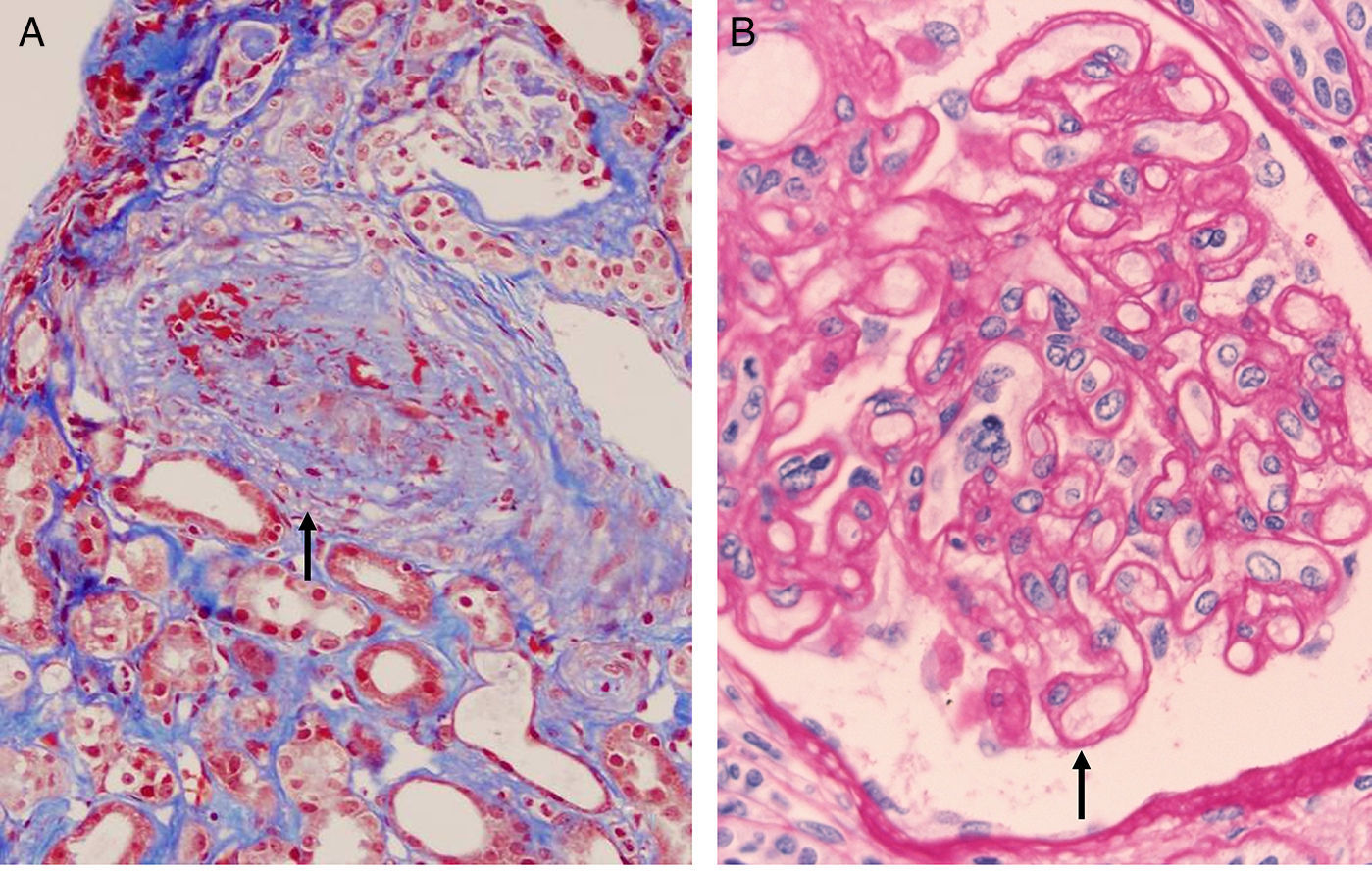
The histology is characterized by:
- 1.
Multilamination and double contour of the basement membrane, mesangial matrix expansion and glomerulitis (light microscopy-PAS and silver staining) (Fig. 8B).
- 2.
Loss of endothelial fenestration, inflammation of endothelial cells and mesangial matrix expansion (electron microscopy).
- 3.
IgM and C3 deposits with positive C4d in varying proportions (immunofluorescence).
Risk factors: patient age, presence of DSA, prior acute rejection and positive C-virus serology.
The pathophysiology involves class I and II DSA, but more often class II, DP as well as DR and DQ. Although having DQ is considered an increased risk of transplant glomerulopathy,75 not all agree with this theory and even claim that there are no differences between DP, DR and DQ DSA.76,77 Why transplant glomerulopathy is related to class II antibodies is unknown.75,78,79
Since about half the patients have no anti-HLA antibodies, the involvement of other etiologies, particularly thrombotic microangiopathy and hepatitis C, have been suggested.80
The role of C4d in the diagnosis of transplant glomerulopathyCurrent data on C4d deposition as a marker of humoral rejection in transplant glomerulopathy is summarized below:
- 1.
The presence of C4d deposits in the glomerulus is useful for diagnosis.81
- 2.
A strong association exists between transplant glomerulopathy and the presence of circulating anti-HLA antibodies and C4d deposition in peritubular capillaries.82
- 3.
In patients with circulating antibodies, C4d deposits can be detected in the glomeruli, but not in the peritubular capillaries. In this case, C4d deposition should be assessed in paraffin sections, since after freezing peripheral C4d deposits can be found in normal glomeruli.83,84
- 4.
Detection of C4d deposits varies according to the series and the technique used. Chronic injury from antibodies occurs in waves and C4d deposition may occur during peak periods.
- 5.
Regarding the problems posed by C4d deposition, the concept of C4d-negative humoral rejection has been proposed for cases in which light microscopy shows glomerulitis and capillaritis.
A plethora of polymorphic non-HLA molecules associated with acute and chronic humoral rejection has been described, but the absence of commercial assays prevents diagnosis. Terasaki suggests that in C4d-positive cases without demonstration of circulating anti-HLA antibodies this possibility should be considered.85
The following non-HLA antibodies should be emphasized:
- 1.
MICA antigens. The polymorphic MHC class I-related chain A (MICA) antigens expressed on endothelial cells have been implicated in the pathogenesis of hyperacute, acute and chronic allograft rejections, although no study involving MICA antibodies has yet demonstrated donor specificity.39
- 2.
Angiotensin II AT1 receptor. In patients with pre-eclampsia with seizures and severe hypertension, agonistic antibodies against the angiotensin II AT1 receptor have been detected in serum.86 Based on these data, in RT recipients with vascular rejection refractory to treatment and with severe hypertension, analysis of the presence of angiotensin II AT1 agonistic antibodies was performed. Of 20 cases, 16 had these IgG antibodies of the subclasses IgG1 and IgG3. In vitro stimulation of vascular cells with AT1-receptor-activating antibody induced phosphorylation of ERK kinase and increased the DNA binding activity of the transcription factors AP-1 and NF-κB, resulting in increased expression of proinflammatory cytokines, procoagulatory genes and cell proliferation. Furthermore, in a renal transplant model in rats, the administration of antibodies against the AT1 angiotensin II receptor caused vasculopathy, which was preventable with losartan.87
- 3.
Vimentin. This protein is part of the intermediate filaments of the intracellular cytoskeleton of the embryonic, blood and endothelial cells of coronary vessels. Vimentin monomers are wound together to form part of the support of the intracellular organelles (mitochondria, endoplasmic reticulum, etc.). It can induce coronary artery disease after cardiac transplantation.88
The authors declare no conflict of interest.
The authors thank the Nephrology, Immunology and Pathology teams from Regional University Hospital (Malaga, Spain) for their collaboration. This study was supported in part by the Spanish Ministry of Economy and Competitiveness (MINECO) (Grant no, ICI14/00016) from the Instituto de Salud Carlos III, RETICS (REDINREN RD 12/0021/0015), and by Grant PI-0590-2012 (in part) from the Consejería de Salud del Gobierno de Andalucía. We also thank Maria Repice and Ian Johnstone for their linguistic assistance in the preparation of the text and doctor Antonio Alonso for his assistance in the preparation of the figures.



