



La nefropatía familiar asociada a hiperuricemia (NFH) es un grupo de nefritis tubulointersticiales crónicas causadas por la alteración del gen de la uromodulina (UMOD), y suponen el 1% de la enfermedad renal crónica avanzada (ERCA) que precisa tratamiento renal sustitutivo (TRS). Presentamos el caso de una familia con una mutación del gen de la UMOD no descrita hasta el momento.
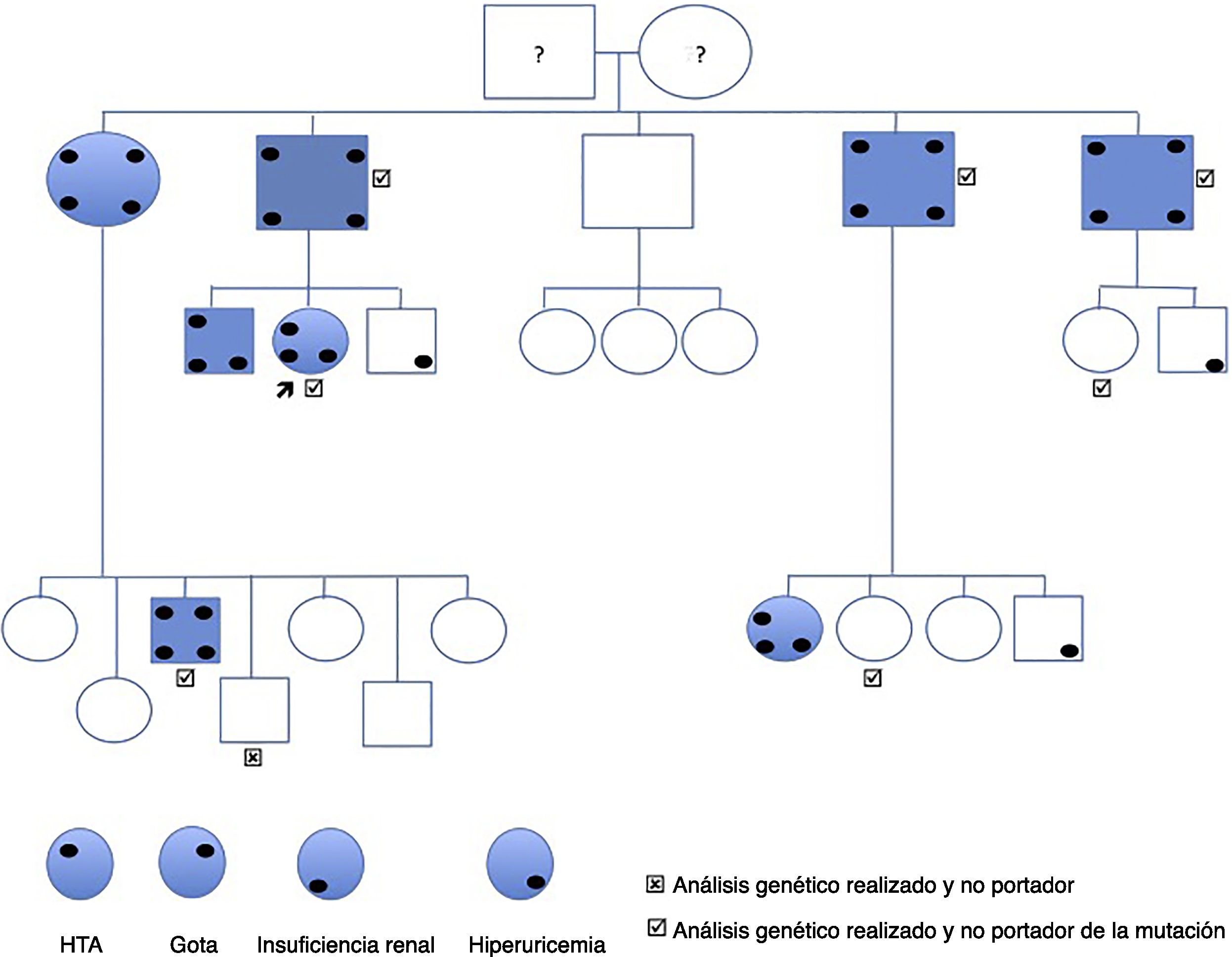
El caso índice es una mujer de 44 años que acude remitida por cifras elevadas de creatinina. Entre sus antecedentes destaca tabaquismo, ulcus bulbar H. pylori+ e hiperuricemia asintomática. No toma medicación habitual y no presenta ninguna clínica extrarrenal asociada. Se diagnostica de HTA grado 1 patrón dipper. Ecografía normal, sin lesiones quísticas. En la analítica destaca hiperuricemia 7,6mg/dl, creatinina 1,26, MDRD 46,8ml/min/1,73m2, orina normal y ClCr (orina 24h) 58ml/min. En su familia, su padre y 3/4 tíos paternos presentan insuficiencia renal diagnosticada en edades tardías y en estado avanzado, uno en estadio 3b y los otros 2 en estadio 4 y 5 (fig. 1). Se valora el resto de familiares de primer y segundo grado y se detecta:
- ?
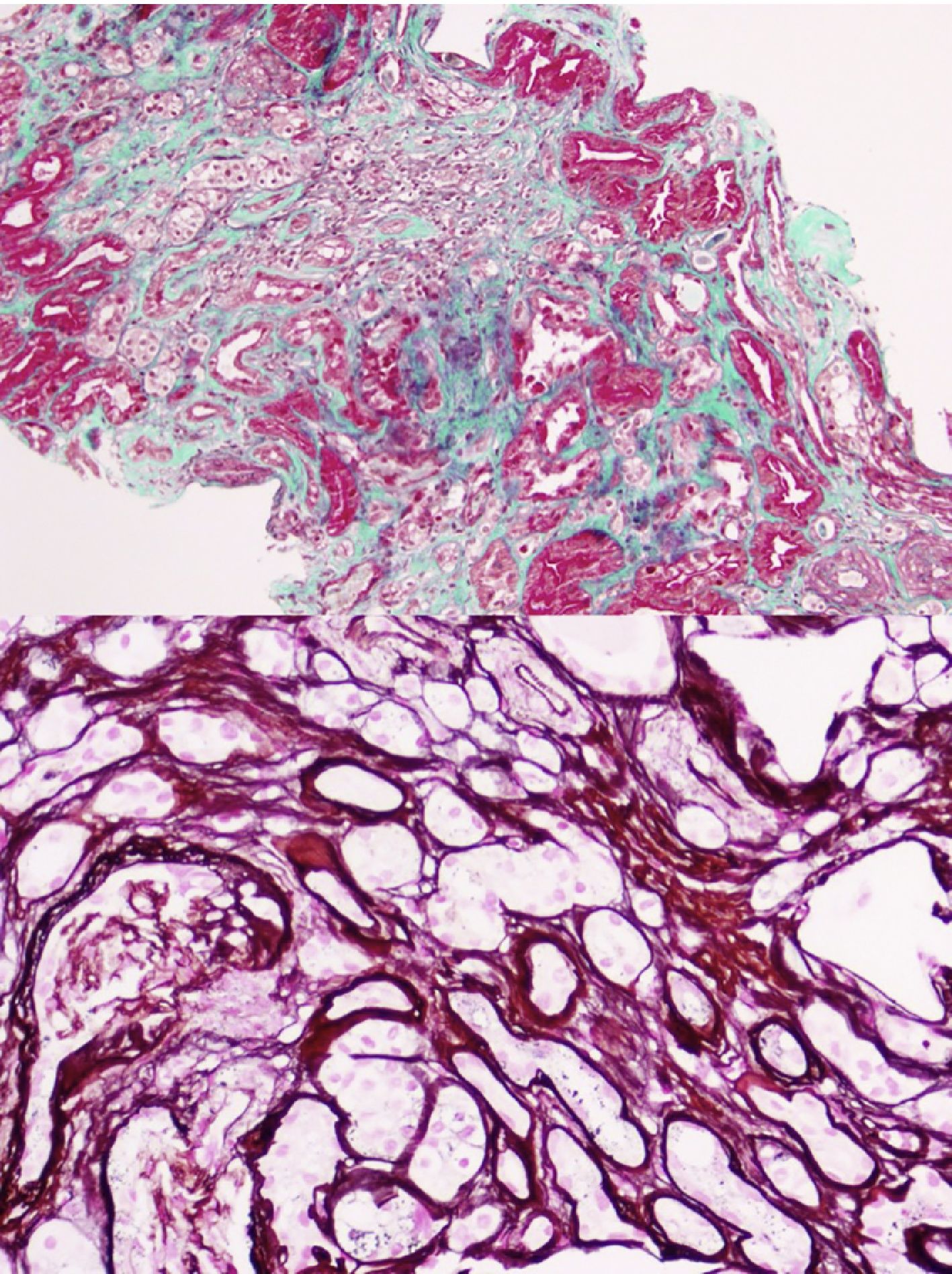
La primera generación consta de 5 personas (padre y 4 tíos de la paciente caso). Todos los que tienen insuficiencia renal también presentan hiperuricemia. En la actualidad, 2 están trasplantados de riñón y 2 con ERCA. En todos ellos menos en uno la ecografía renal es normal sin evidencia de lesiones quísticas. Se realiza biopsia renal en el único tío paterno con solo un quiste corticomedular y con ERCA MDRD-4 20ml/min/1,73m2, sin poder apreciar más que nefropatía crónica tubulointersticial (fig. 2).
- ?
La segunda generación consta de la paciente caso, 2 hermanos y 16 primos. Se detecta insuficiencia renal en 4 casos incluyendo a la paciente, 3 en estadio 3a y uno en estadio 2, todos ellos con hiperuricemia, y 3 casos con hiperuricemia sin insuficiencia renal. Solo una prima, sin insuficiencia renal ni hiperuricemia presenta quistes corticomedulares.
- ?
La tercera generación consta de 34 personas nacidas entre 1981 y 2013, todas estudiadas con analítica de sangre y orina normales.
Globalmente, encontramos:
- ?
Insuficiencia renal en 4/5 ascendentes, 1/2 hermanos y 2 primos hermanos.
- ?
Hiperuricemia con hipouricosuria en todos aquellos que presentan insuficiencia renal. Además, osmolaridad urinaria<500mOsm/kg e hipomagnesuria con normomagnesemia en algunos.
- ?
La presencia de quistes corticomedulares solo se encontró en 2 de los familiares estudiados, uno con afectación y otro sin afectación clínico-analítica evidente. Ninguno mostró clínica o ecográficamente litiasis renal.
- ?
Gota solo en los 4 afectados de la primera generación.
- ?
La HTA coincide en todos los casos con la insuficiencia renal.
Una vez estudiado clínicamente a la familia se realizó una secuenciación de los exones 2 a 12 (codificantes), así como las bases intrónicas flanqueantes en uno de los afectados. Para ello se amplificaron fragmentos mediante PCR seguido de secuenciación automática mediante el método de Sanger. Identificamos una mutación nueva en el exón 4: c.517C>G, p.P173A, que se determinó en todos los miembros de la familia genéticamente estudiados (fig. 1), siendo todos los afectados portadores heterocigotos. El familiar que no presenta la mutación tampoco presenta enfermedad relacionada clínica ni analítica. De los otros 6 que sí presentan la mutación, 4 han desarrollado la enfermedad (hiperuricemia e insuficiencia renal), mientras que 2 no.
Por todo ello se diagnostica de nefropatía intersticial crónica familiar con hiperuricemia causada por el gen de la UMOD, variante enfermedad quística medular tipo 2.
Mutaciones en el gen de la UMOD dan lugar a diferentes nefropatías hereditarias hiperuricemiantes e hipouricosúricas que pueden llegar a provocar, además de gota y litiasis, hipertensión arterial y nefritis tubulointersticial crónica que conduce a insuficiencia renal crónica avanzada1,2. La NFH de herencia autonómica dominante suele manifestarse en la edad adulta por hiperuricemia, existiendo una gran heterogeneidad genotipo-fenotipo intra e interfamiliar de esta entidad, situación que también ocurre en la familia expuesta3,4.
Se han descrito decenas de mutaciones en el gen de la UMOD5,6. En nuestro caso se ha descubierto una mutación no descrita previamente que trata de una modificación del aminoácido citosina por guanina (c.517C>G p.pro173Ala) en el fragmento estudiado correspondiente al exón 3 del gen de la UMOD, y es por ello que lo ponemos en conocimiento de la comunidad científica. La mayoría de las mutaciones se han localizado en el exón 47, pero nunca la mutación actual que condiciona el cambio de aminoácido arriba descrito. La novedad de esta mutación no implicaría carácter patogénico de no ser por haberse encontrado en varios miembros afectos de la familia (fig. 2). Este hallazgo ha permitido el diagnóstico preclínico de 2 familiares de 39 y 40 años, y su descarte en un individuo. Dado que todos los afectados tienen la mutación, pero hay portadores sin afectación, entendemos que la mutación se relaciona con una penetrancia variable, que podría depender de la edad y es por ello que, aunque la progresión de la insuficiencia renal en esta entidad es lenta, su carácter silente justifica la monitorización de los familiares jóvenes asintomáticos, ya que, aunque no exista una terapia específica, el diagnóstico precoz nos permitiría el tratamiento temprano de la hiperuricemia indolente y recomendaciones globales de nefroprotección que conduzcan a un enlentecimiento en la aparición o progresión de la nefropatía8-10.
Concluimos que la detección precoz de la NFH puede conducir a un tratamiento temprano que retrase la aparición o progresión de la insuficiencia renal. El estudio familiar y análisis genético en esta enfermedad son importantes para su diagnóstico definitivo ya que la clínica y la histopatología no son específicas.



