


La rama gruesa ascendente del asa de Henle (TAL) reabsorbe aproximadamente el 30% de NaCl filtrado mediante 2 mecanismos: reabsorción transepitelial y paracelular. Esta última se efectúa a través de un tipo de proteínas de las uniones estrechas conocidas como claudinas. La mutación en el gen que codifica la claudina 10 ocasiona un raro trastorno tubular, pierde sal, con alcalosis metabólica hipopotasémica, pero que a diferencia del síndrome de Bartter y enfermedad de Gitelman, suele cursar con hipermagnesemia y con manifestaciones extrarrenales como xerostomía, alacrimia e hipohidrosis con ictiosis conocido con el acrónimo de síndrome de HELIX.
The thick ascending limb of the loop of Henle (TAL) reabsorbs approximately 30% of filtered NaCl through two mechanisms: transepithelial and paracellular reabsorption. The latter is carried out through a class of tight junction proteins known as claudins. A mutation in the gene encoding claudin-10 causes a rare salt-wasting tubular disorder with hypokalemic metabolic alkalosis. However, unlike Bartter syndrome and Gitelman disease, it usually presents with hypermagnesemia and extrarenal manifestations such as xerostomia, alacrima, and hypohidrosis with ichthyosis, known by the acronym HELIX syndrome.
La rama gruesa ascendente del asa de Henle (TAL) juega un papel fundamental en la fisiología del riñón humano participando en la reabsorción de sodio, en los mecanismos de concentración de la orina, en la homeostasis del calcio, magnesio, bicarbonato y amonio, y en la síntesis de uromodulina regulando la composición de proteínas urinarias1.
La TAL reabsorbe aproximadamente el 30% del NaCl filtrado mediante 2 mecanismos, reabsorción transepitelial y paracelular. La reabsorción transepitelial depende de la acción conjunta de 2 proteínas apicales (cotransportador NKCC2 (Na+-K+-2Cl–) y canal de potasio Ki1.1) y de 4 proteínas basolaterales (Na-K-ATPasa, canales de cloro CLC-Ka y ClC-Kb, y su subunidad bartina)2,3. La vía paracelular, responsable de la reabsorción de casi el 50% del sodio, es promovida por un potencial transepitelial luz positiva y regulada por un tipo de claudina, la claudina-10b1,4. Las claudinas son una familia de proteínas transmembrana integrales que forman parte de las uniones estrechas de las células epiteliales y endoteliales, y desempeñan un papel crucial en el control del transporte paracelular de iones, agua y otras moléculas pequeñas. En el riñón, cada segmento tubular expresa un conjunto específico de claudinas que confieren propiedades únicas en cuanto a permeabilidad y selectividad de la vía paracelular además de contribuir a mantener la polaridad celular4.
Las claudinopatías son trastornos hereditarios causados por mutaciones en los genes que codifican las claudinas, dando lugar a enfermedades conocidas como el síndrome de hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (claudinas 16 y 19)5, sordera neurosensorial autosómica recesiva no sindrómica (claudina 14)6, así como ictiosis-colangitis esclerosante neonatal (claudina 1)7. Determinados polimorfismos en el gen que codifica la claudina 14, mediante su interacción con la claudina 16, se han asociado al desarrollo de hipercalciuria y cálculos renales8.
La mayoría de los trastornos tubulares pierde sal con activación del eje renina-aldosterona que afectan a la rama gruesa ascendente del asa del Henle y al túbulo contorneado distal (DCT), tales como el síndrome de Bartter y enfermedad de Gitelman, respectivamente, cursan con alcalosis metabólica hipopotasémica e hipomagnesemia. A continuación, se describe el caso de un paciente con un fenotipo de tubulopatía pierde sal, pero con el hallazgo característico de niveles elevados de magnesio en plasma.
Reporte de un casoVarón de 2 años de edad, origen Colombia, remitido a la consulta de nefrología infantil, por hallazgo accidental confirmado de una creatinina elevada (Cr: 0,47mg/dl) detectada en el contexto de un estudio de retraso del desarrollo psicomotor. No consanguinidad referida. No antecedentes familiares contributorios salvo un abuelo materno fallecido por enfermedad renal crónica de etiología no filiada. Entre los antecedentes personales destaca una hidrocefalia benigna externa diagnosticada por resonancia magnética cerebral y una xerosis cutánea con ligera ictiosis descamativa, anhidrosis y disminución de la producción de saliva controlada por dermatología, sin desgaste del esmalte dental ni inflamación gingival. No presenta retraso pondero-estatural (peso: 11,5kg [Zscore −0,48]; talla: 87cm [Zscore −0,26]).
Desde el punto de vista clínico, el paciente describe un cuadro de poliuria-polidipsia sin referir otras anomalías clínicas. No consumo de fármacos nefrotóxicos. Ausencia de hematuria macroscópica. Sin antecedentes de infección del tracto urinario.
Hallazgos de laboratorio: Función renal: urea 45mg/dl, creatinina 0,47mg/dl, FG (Schwartz_09): 76ml/min/1,73m2, Na 135mEq/l, K 3,8mEq/l, Cl 98mEq/l, Ca 8,9mg/dl, P 6,3mg/dl, Mg 4,8mg/dl, úrico 5,4mg/dl, EFNa 0,9%, RTP 90%, EFK 21,37%, EFMg 1,98%, cociente Ca/Cr: 0,02mg/mg, pH 7,42, bicarbonato 26,6mmol/l, EB 1,9mmol/l. Ausencia de anomalías en el sedimento urinario. Test de concentración urinaria tras desmopresina: 661mOsm/kg. Normotenso. Ecografía renal sin hallazgos patológicos, ausencia de nefrocalcinosis.
Ante la sospecha diagnóstica de enfermedad renal crónica de probable etiología túbulo-intersticial se solicita estudio genético con panel de secuenciación de exoma dirigido a trastornos tubulares y nefritis túbulo-intersticial autosómica dominante (NTIAD) con resultado negativo.
Durante su seguimiento en consultas externas, progresivamente se objetiva una tubulopatía pierde sal con un fenotipo de enfermedad de Gitelman con activación del sistema renina-angiotensina-aldosterona (renina: 467μU/ml y aldosterona 621pg/ml), alcalosis metabólica hipopotasémica (pH 7,41, bicarbonato 26mmol/l, K 2,57mEq/l) e hipocalciuria (0,02mg/mg), pero a diferencia de la anterior, con hipermagnesemia (5,1mg/dl) sin referir sintomatología sugestiva de la misma. Ante el cuadro descrito junto con las manifestaciones extrarrenales de xerosis con ictiosis descamativa y anhidrosis, se solicita estudio genético de claudinopatías detectando una variante patogénica (variante c.316_323delGGCTCCGA, p.(Gly106*) en el exón 3a del gen CLDN-10b en aparente homocigosis compatible con síndrome de HELIX.
Se recomienda estudio de segregación familiar, pero únicamente da su consentimiento la madre, en cuyo análisis no se detecta la variante estudiada, lo que sugiere la aparición de una mutación de novo en el paciente sin haber podido excluir la existencia de consanguinidad en los progenitores.
En cuanto al tratamiento requiere suplementos de sodio (cloruro sódico: 3mEq/kg) y potasio (cloruro potásico: 2,5mEq/kg), así como abundante ingesta hídrica. No requiere tratamiento de la hipermagnesemia.
Actualmente, a la edad de 8 años, presenta una adecuada curva pondero-estatural con FG (Schwartz_09): 81ml/min/1,73m2 y las alteraciones electrolíticas descritas previamente. También presenta leve retraso global del desarrollo en seguimiento por neuropediatría y xerosis y anhidrosis en seguimiento por dermatología.
DiscusiónEl síndrome de Hypohidrosis, Electrolyte disturbances, Lacrimal gland dysfunction, Ichthyosis, and Xerostomia (HELIX) es un raro trastorno tubular hereditario, pierde sal, con manifestaciones renales y extrarrenales acuñado por Hadj-Rabia et al en 20189,10, aunque previamente descrito como un nuevo trastorno tubular en 2017 por Bongers et al.11.
Las manifestaciones desde el punto de vista renal, son consecuencia de una reducción de la permeabilidad paracelular del sodio en la porción gruesa ascendente del asa de Henle dando lugar a un cuadro de poliuria-polidipsia por pérdida hidrosalina, con un defecto de la capacidad de concentración urinaria y alteraciones electrolíticas, que abarca una alcalosis metabólica hipopotasémica por un hiperaldosteronismo secundario con una tendencia a la presión arterial baja, hipermagnesemia, tendencia a la hipercalcemia con hipocalciuria y una lenta progresión a enfermedad renal crónica1,9–11.
Las manifestaciones extrarrenales, sin embargo, afectan a las glándulas exocrinas y se caracterizan por xerostomía con reducción grave del componente acuoso de la saliva, desgaste severo del esmalte, alacrimia, hipohidrosis con intolerancia al calor e ictiosis1,9–11.
Se trata de un trastorno hereditario autosómico recesivo que requiere para su presentación de una mutación en homocigosis o una heterocigosis compuesta en el gen de la claudina 10 localizado en el cromosoma 13 (13q.31.-q.34)9–11. Este gen contiene 6 exones y da lugar a 2 isoformas: claudina-10a y claudina-10b que difieren en el exón 112. Las mutaciones descritas pueden afectar a ambas isoformas como a la claudina-10b en solitario, sin encontrarse diferencias en sus manifestaciones clínicas12. La claudina-10a, que actúa como canal paracelular de aniones (Cl–), está restringida al útero y al riñón, específicamente en el túbulo contorneado proximal13,14. La claudina-10b, por su parte, es la única claudina presente en la franja interna de la médula externa (ISOM), sin embargo, en la franja externa de la médula externa (OSOM) y en la corteza, las claudinas tienen un patrón de expresión en mosaico con uniones estrechas que expresan claudina-10b y claudinas 3, 14, 16 y 19, responsable de la reabsorción de cationes como el Ca2+ y el Mg2+1,9,15. La claudina-10b se caracteriza por ser un canal impermeable al agua pero altamente permeable al Na+ contribuyendo a disipar el potencial transepitelial positivo en la luz tubular de la TAL4,12,14,15. Además de estar presente en el riñón, la claudina-10b también se localiza en glándulas salivales, glándulas sudoríparas, cerebro, pulmones y páncreas4,12.
Breiderhoff et al. en 2012 estudiaron en ratones un modelo sin expresión de claudina 10 en el asa de Henle objetivando una reducción de la selectividad paracelular de Na+ en el TAL que condujo a un defecto de la concentración urinaria con poliuria, polidipsia y niveles elevados de urea acompañado de un incremento de la secreción de K+ y H+ e hipermagnesemia. Estas alteraciones electrolíticas se acompañaban de una nefrocalcinosis medular severa16.
Desde el punto de vista fisiopatológico, la pérdida de función de claudina-10b reduce la reabsorción de Na+ en el TAL por el componente paracelular, incrementando el potencial transepitelial luz positiva que favorece la reabsorción paracelular de Ca2+ y Mg2+ en la porción gruesa ascendente del asa de Henle a través de una redistribución de la claudina 16 y claudina 19 en los segmentos del túbulo1,12,16,17 (fig. 1). Como ocurre con otras tubulopatías pierde sal que afectan a la porción ascendente del asa gruesa, como en el síndrome de Bartter, la activación del sistema renina angiotensina aldosterona así como el aporte distal de Na+ favorece la reabsorción de Na+ a través del receptor sensible a amilorida (ENaC), regulado por la aldosterona, de las células principales así como la secreción de K+ y H+ por las células intercaladas del segmento distal de la nefrona conduciendo al trastorno electrolítico característico de alcalosis metabólica hipopotasémica. Por otro lado, en el epitelio de la glándula salivales, sudoríparas y lacrimales, Na+ y Cl− se secretan a través del cotransportador basolateral NKCC1 y el canal de cloruro apical CFTR, creando un diferencial de potencial transepitelial de luz negativa que permite la secreción pasiva paracelular de Na+ a través de la claudina-10b que actúa como un canal. Esto se asocia con una secreción de agua transcelular a través de los canales de agua de aquaporina-5. La pérdida de función de claudina-10b resulta en un defecto en la secreción de Na+ y agua con las manifestaciones extrarrenales previamente descritas1,12.
, el reciclaje del K+ en la membrana apical (kir1.1) y la vuelta de Na+ del espacio intersticial a la luz tubular a través de uniones estrechas intercelulares contribuyen al desarrollo de un gradiente eléctrico transepitelial luz positiva, que favorece el paso paracelular del Ca2+ y el Mg2+ a través de las claudinas-16 y claudinas-19. El Cl− atraviesa la membrana basolateral a través del transportador ClC-Ka y ClC-Kb mientras que el Na+ la atraviesa a través de la bomba de Na-K ATPasa acoplado a la entrada de K+ al interior celular. De esta manera se genera un gradiente químico necesario para la reabsorción de Na+ a través del cotransportador NKCC2. Determinadas células del asa de Henle expresan una proteína Claudina-10b responsable de la reabsorción paracelular de Na+ disipando el gradiente eléctrico transepitelial luz positiva. Su disfunción, por tanto, reduce la reabsorción paracelular de Na+, incrementando el potencial transepitelial luz positiva, responsable del aumento de la reabsorción de Ca2+ y Mg2+ a través de una redistribución de la claudina-16 y claudina-19. Ca2+: calcio; Cl−: cloro; K+: potasio; Mg2+: magnesio; Na+: sodio.")
Modelo especulativo de la fisiopatología del síndrome de Helix en la porción gruesa ascendente del asa de Henle. La reabsorción de Na+, 2Cl− y K+ a través del transportador sensible a la furosemida (NKCC2), el reciclaje del K+ en la membrana apical (kir1.1) y la vuelta de Na+ del espacio intersticial a la luz tubular a través de uniones estrechas intercelulares contribuyen al desarrollo de un gradiente eléctrico transepitelial luz positiva, que favorece el paso paracelular del Ca2+ y el Mg2+ a través de las claudinas-16 y claudinas-19. El Cl− atraviesa la membrana basolateral a través del transportador ClC-Ka y ClC-Kb mientras que el Na+ la atraviesa a través de la bomba de Na-K ATPasa acoplado a la entrada de K+ al interior celular. De esta manera se genera un gradiente químico necesario para la reabsorción de Na+ a través del cotransportador NKCC2. Determinadas células del asa de Henle expresan una proteína Claudina-10b responsable de la reabsorción paracelular de Na+ disipando el gradiente eléctrico transepitelial luz positiva. Su disfunción, por tanto, reduce la reabsorción paracelular de Na+, incrementando el potencial transepitelial luz positiva, responsable del aumento de la reabsorción de Ca2+ y Mg2+ a través de una redistribución de la claudina-16 y claudina-19. Ca2+: calcio; Cl−: cloro; K+: potasio; Mg2+: magnesio; Na+: sodio.
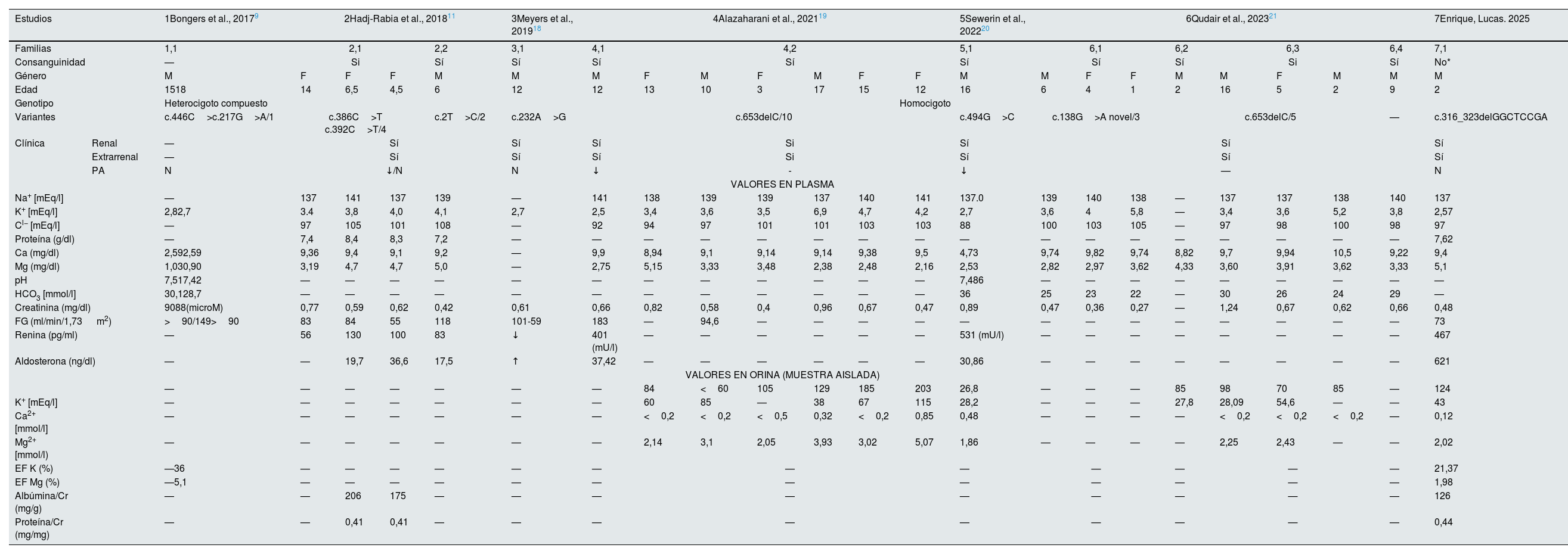
En la actualidad se han descrito 37 casos de 11 familias9,18–22. Esta publicación es la primera que describe las características de los pacientes pediátricos con este síndrome hasta la fecha. En la tabla 1 se analizan los principales datos genéticos, clínicos y analíticos de los pacientes pediátricos, incluyendo nuestro paciente. El rango de edades oscila desde un año hasta los 18 años. Dado el tipo de herencia de esta enfermedad, la mayoría de los pacientes tienen antecedentes de consanguinidad a excepción de nuestro paciente que no se pudo confirmar. Todos los pacientes, a excepción de uno, tienen una mutación «missense» en homocigosis. Desde el punto de vista clínico, en todos los pacientes se describen las manifestaciones extrarrenales propias de la enfermedad, aunque hay que destacar heterogeneidad en su gravedad. Aunque no existen datos descritos relativos a la capacidad de concentración urinaria, es relativamente frecuente un defecto de la capacidad de concentración que puede deberse a la pérdida salina que impide crear una adecuada tonicidad del intersticio medular, así como a la hipopotasemia crónica. La hipermagnesemia es un hallazgo común en todos los pacientes pediátricos disminuyendo su frecuencia con la edad. En una proporción importante de pacientes los valores de potasio se encontraban en rangos de normalidad, no obstante, es necesario destacar que la hipopotasemia es más frecuente en los adultos que en los niños probablemente como consecuencia de mecanismo de compensación que existen en los niños a pesar de la activación del eje renina-aldosterona. En los pacientes en el que el equilibrio ácido-base fue estudiado, se objetiva una alcalosis metabólica con presión arterial baja e hipocalciuria, tal y como describimos en nuestro paciente. La frecuencia de enfermedad renal crónica descritas en los adultos es hasta de un 25% de los casos12,18, sin embargo, es un hallazgo relativamente frecuente en los pacientes pediátricos estudiados. El mecanismo patogénico subyacente en los adultos podría estar relacionado con inflamación del intersticio con vasoconstricción y aumento de la fibrosis secundaria a la hipopotasemia crónica23,24, sin embargo, en los niños se requieren más estudios. La albuminuria también es un hallazgo descrito en algunos pacientes de la serie como nuestro caso. En ecografía renal no se detectan alteraciones ni en el tamaño ni en la ecogenicidad de los riñones, así como una ausencia de nefrocalcinosis, al contrario de lo que sucede en modelo de ratones estudiado12,16.
Datos demográficos, genéticos, clínicos y analíticos de los pacientes en edad pediátrica diagnosticados de síndrome de HELIX
| Estudios | 1Bongers et al., 20179 | 2Hadj-Rabia et al., 201811 | 3Meyers et al., 201918 | 4Alazaharani et al., 202119 | 5Sewerin et al., 202220 | 6Qudair et al., 202321 | 7Enrique, Lucas. 2025 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Familias | 1,1 | 2,1 | 2,2 | 3,1 | 4,1 | 4,2 | 5,1 | 6,1 | 6,2 | 6,3 | 6,4 | 7,1 | ||||||||||||
| Consanguinidad | — | Si | Sí | Sí | Sí | Sí | Sí | Sí | Sí | Si | Sí | No* | ||||||||||||
| Género | M | F | F | F | M | M | M | F | M | F | M | F | F | M | M | F | F | M | M | F | M | M | M | |
| Edad | 1518 | 14 | 6,5 | 4,5 | 6 | 12 | 12 | 13 | 10 | 3 | 17 | 15 | 12 | 16 | 6 | 4 | 1 | 2 | 16 | 5 | 2 | 9 | 2 | |
| Genotipo | Heterocigoto compuesto | Homocigoto | ||||||||||||||||||||||
| Variantes | c.446C>c.217G>A/1 | c.386C>T c.392C>T/4 | c.2T>C/2 | c.232A>G | c.653delC/10 | c.494G>C | c.138G>A novel/3 | c.653delC/5 | — | c.316_323delGGCTCCGA | ||||||||||||||
| Clínica | Renal | — | Sí | Sí | Sí | Si | Sí | Sí | Sí | |||||||||||||||
| Extrarrenal | — | Sí | Sí | Sí | Si | Sí | Sí | Sí | ||||||||||||||||
| PA | N | ↓/N | N | ↓ | - | ↓ | — | N | ||||||||||||||||
| VALORES EN PLASMA | ||||||||||||||||||||||||
| Na+ [mEq/l] | — | 137 | 141 | 137 | 139 | — | 141 | 138 | 139 | 139 | 137 | 140 | 141 | 137.0 | 139 | 140 | 138 | — | 137 | 137 | 138 | 140 | 137 | |
| K+ [mEq/l] | 2,82,7 | 3.4 | 3,8 | 4,0 | 4,1 | 2,7 | 2,5 | 3,4 | 3,6 | 3,5 | 6,9 | 4,7 | 4,2 | 2,7 | 3,6 | 4 | 5,8 | — | 3,4 | 3,6 | 5,2 | 3,8 | 2,57 | |
| Cl− [mEq/l] | — | 97 | 105 | 101 | 108 | — | 92 | 94 | 97 | 101 | 101 | 103 | 103 | 88 | 100 | 103 | 105 | — | 97 | 98 | 100 | 98 | 97 | |
| Proteína (g/dl) | — | 7,4 | 8,4 | 8,3 | 7,2 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 7,62 | |
| Ca (mg/dl) | 2,592,59 | 9,36 | 9,4 | 9,1 | 9,2 | — | 9,9 | 8,94 | 9,1 | 9,14 | 9,14 | 9,38 | 9,5 | 4,73 | 9,74 | 9,82 | 9,74 | 8,82 | 9,7 | 9,94 | 10,5 | 9,22 | 9,4 | |
| Mg (mg/dl) | 1,030,90 | 3,19 | 4,7 | 4,7 | 5,0 | — | 2,75 | 5,15 | 3,33 | 3,48 | 2,38 | 2,48 | 2,16 | 2,53 | 2,82 | 2,97 | 3,62 | 4,33 | 3,60 | 3,91 | 3,62 | 3,33 | 5,1 | |
| pH | 7,517,42 | — | — | — | — | — | — | — | — | — | — | — | — | 7,486 | — | — | — | — | — | — | — | — | — | |
| HCO3 [mmol/l] | 30,128,7 | — | — | — | — | — | — | — | — | — | — | — | — | 36 | 25 | 23 | 22 | — | 30 | 26 | 24 | 29 | — | |
| Creatinina (mg/dl) | 9088(microM) | 0,77 | 0,59 | 0,62 | 0,42 | 0,61 | 0,66 | 0,82 | 0,58 | 0,4 | 0,96 | 0,67 | 0,47 | 0,89 | 0,47 | 0,36 | 0,27 | — | 1,24 | 0,67 | 0,62 | 0,66 | 0,48 | |
| FG (ml/min/1,73 m2) | >90/149>90 | 83 | 84 | 55 | 118 | 101-59 | 183 | — | 94,6 | — | — | — | — | — | — | — | — | — | — | — | — | — | 73 | |
| Renina (pg/ml) | — | 56 | 130 | 100 | 83 | ↓ | 401 (mU/l) | — | — | — | — | — | — | 531 (mU/l) | — | — | — | — | — | — | — | — | 467 | |
| Aldosterona (ng/dl) | — | — | 19,7 | 36,6 | 17,5 | ↑ | 37,42 | — | — | — | — | — | — | 30,86 | — | — | — | — | — | — | — | — | 621 | |
| VALORES EN ORINA (MUESTRA AISLADA) | ||||||||||||||||||||||||
| — | — | — | — | — | — | — | 84 | <60 | 105 | 129 | 185 | 203 | 26,8 | — | — | — | 85 | 98 | 70 | 85 | — | 124 | ||
| K+ [mEq/l] | — | — | — | — | — | — | — | 60 | 85 | — | 38 | 67 | 115 | 28,2 | — | — | — | 27,8 | 28,09 | 54,6 | — | — | 43 | |
| Ca2+ [mmol/l] | — | — | — | — | — | — | — | <0,2 | <0,2 | <0,5 | 0,32 | <0,2 | 0,85 | 0,48 | — | — | — | — | <0,2 | <0,2 | <0,2 | — | 0,12 | |
| Mg2+ [mmol/l) | — | — | — | — | — | — | — | 2,14 | 3,1 | 2,05 | 3,93 | 3,02 | 5,07 | 1,86 | — | — | — | — | 2,25 | 2,43 | — | — | 2,02 | |
| EF K (%) | —36 | — | — | — | — | — | — | — | — | — | — | — | — | 21,37 | ||||||||||
| EF Mg (%) | —5,1 | — | — | — | — | — | — | — | — | — | — | — | — | 1,98 | ||||||||||
| Albúmina/Cr (mg/g) | — | — | 206 | 175 | — | — | — | — | — | — | — | — | — | 126 | ||||||||||
| Proteína/Cr (mg/mg) | — | — | 0,41 | 0,41 | — | — | — | — | — | — | — | — | — | 0,44 | ||||||||||
Los estudios se representan en la primera fila en orden cronológico (1 - Bongers et al., 20179; 2 - Hadj-Rabia et al., 201811; 3 - Meyers et al., 201918; 4 - Alzaharani et al., 202119; 5 - Sewerin et al., 202220; 6 - Qudair et al., 202321; 7 - Enrique, Lucas. 2025). Las familias de cada estudio se representan en la segunda fila. F: femenino; M: masculino; (—): no disponible. PA: presión arterial. N=normal; ↓=bajo; ↑: alto; No*: no es posible descartar consanguinidad. Ca2+: calcio; Cl−: cloro; Cr: creatinina; EF Mg: excreción fraccional de magnesio; FG: filtrado glomerular; HCO3: bicarbonato; K+: potasio; Mg2+: magnesio; Na+: sodio.
El diagnóstico definitivo de este síndrome se realiza mediante estudio genético con secuenciación del gen descrito, imprescindible para su consejo genético. No requiere biopsia renal.
No existe un tratamiento específico para mejorar el estado de los pacientes con síndrome HELIX. Se recomienda una ingesta elevada de NaCl y líquidos, con suplementos de potasio y fármacos que bloqueen la secreción y/o la acción de la aldosterona o bloqueadores del canal epitelial de Na+ (ENaC) cuando hay hipopotasemia refractaria. Se pueden utilizar lágrimas artificiales y saliva para aliviar los síntomas de sequedad ocular y bucal, respectivamente. Se debe desaconsejar la actividad física intensa prolongada, en particular cuando la temperatura exterior es alta, para prevenir el riesgo de hipertermia.
Conflicto de interesesLos autores no presentan conflictos de intereses.



