






About 80% of patients with tuberous sclerosis complex (TSC) present renal involvement, usually as angiomyolipomas followed by cystic disease. An early diagnosis of polycystic kidney disease (PKD) in such patients is frequently related to the TSC2/PKD1 contiguous gene syndrome (PKDTS). Molecular confirmation of PKDTS is important for a prompt diagnosis, which can be complicated by the phenotypic heterogeneity of PKD and the absence of a clear phenotype–genotype correlation. Herein, we report three PKDTS pediatric patients. The case 3 did not present a classic PKDTS phenotype, having only one observable cyst on renal ultrasound at age 4 and multiple small cysts on magnetic resonance imaging at age 15. In this patient, chromosomal microarray analysis showed a gross deletion of 230.8kb that involved TSC2, PKD1 and 13 other protein-coding genes, plus a heterozygous duplication of a previously undescribed copy number variant of 242.9kb that involved six protein-coding genes, including SSTR5, in the 16p13.3 region. Given the observations that the case 3 presented the mildest renal phenotype, harbored three copies of SSTR5, and the reported inhibition of cystogenesis (specially in liver) observed with somatostatin analogs in some patients with autosomal dominant PKD, it can be hypothesized that other genetic factors as the gene dosage of SSTR5 may influence the PKD phenotype and the progression of the disease; however, future work is needed to examine this possibility.
Un 80% de los pacientes con complejo de esclerosis tuberosa (CET) presentan afectación renal, generalmente angiomiolipomas, seguidos de enfermedad quística. Un diagnóstico temprano de la enfermedad renal poliquística (ERP) en estos pacientes se relaciona con frecuencia con el síndrome de genes contiguos TSC2/PKD1 (PKDTS). La confirmación molecular de PKDTS es importante para establecer un diagnóstico oportuno, que puede complicarse por la heterogeneidad fenotípica de PKD y la ausencia de una clara correlación entre fenotipo y genotipo. En este artículo presentamos los casos de 3 pacientes pediátricos con PKDTS. El caso 3 no presentó un fenotipo PKDTS clásico, con solo un quiste observable en la ecografía renal a los 4 años y numerosos quistes pequeños en la resonancia magnética a los 15 años. En este paciente, el análisis de microarreglos para análisis cromosómico global mostró una eliminación total de 230,8kb que involucró a TSC2,PKD1 y otros 13 genes codificantes de proteínas, más una duplicación heterocigota para una variante de número de copias no descrita previamente de 242,9kb que involucró a 6 genes codificantes de proteínas, entre ellos SSTR5, en la región 16p13.3. Dado que el caso 3 mostraba el fenotipo renal menos severo, contaba con tres copias del gen SSTR5 y a que se ha observado una inhibición en la cistogénesis (especialmente en el hígado) con los análogos de somatostatina en algunos pacientes con ERP autosómica dominante, podemos hipotetizar que existen otros factores genéticos como la dosis génica de SSTR5 que pudieran influir en el fenotipo y la progresión de la ERP; sin embargo, se necesitan estudios adicionales para investigar esta posibilidad.
Tuberous sclerosis complex (TSC, MIM#191100) is an autosomal dominant disorder that is characterized by the development of multiple hamartomas and is caused by heterozygous pathogenic variants in the tumor suppressor genes, TSC1 or TSC2. The most common renal lesions are angiomyolipomas, occurring in >80% of the patients and frequently presented as multiple and bilateral. Besides, approximately 14–32% of the TSC cases exhibit some degree of renal cyst formation, most of which occurs in the second decade of life.1 Early development of multiple renal cysts with kidney enlargement in TSC patients is associated with the heterozygous contiguous deletion of TSC2 and the adjacent PKD1 gene; this last is the causative for autosomal dominant polycystic kidney disease (ADPKD, MIM#173900). The TSC2/PKD1 contiguous gene syndrome (PKDTS, MIM #600273) reportedly comprises ∼2–5% of all TSC cases.2,3 These patients should be diagnosed as early as possible due to the PKD1 deletion that increases the risk for ADPKD-related complications such as cystic kidney disease, hepatic and pancreatic cysts, arterial hypertension, intracranial aneurysm3–5 and to the probability of presenting an early end-stage renal disease (20–30 years).6
Molecular analysis is crucial for the early and confirmed diagnosis of PKDTS, which allows clinicians to assess for the progressive renal lesions and other potentially fatal ADPKD-related complications.7 To date, more than 65 molecularly characterized cases of PKDTS have been reported (Fig. 1).4,8–16 However, the precise deletion interval has only been identified in a few of these patients, and no clear genotype–phenotype correlation has been established. Hence, we need to describe more PKDTS patients and their responsible genotypes in order to identify possible genotype–phenotype correlations and/or the mechanisms underlying the phenotypic variability of PKDTS.
.")
TSC2 and PKD1 deletions characterized by various molecular techniques in the reported PKDTS cases.
, Complete deletion of the gene; , partial deletion of the gene; ?, Unknown breakpoint. Abbreviations: aCGH: microarray-based comparative genomic hybridization; CMA: chromosomal microarray; FISH: fluorescence in situ hybridization; MLPA: multiplex ligation-dependent probe amplification; qPCR: quantitative PCR, *Cases in which PKD symptoms and end-stage renal disease were recognized fairly late (>20 years).

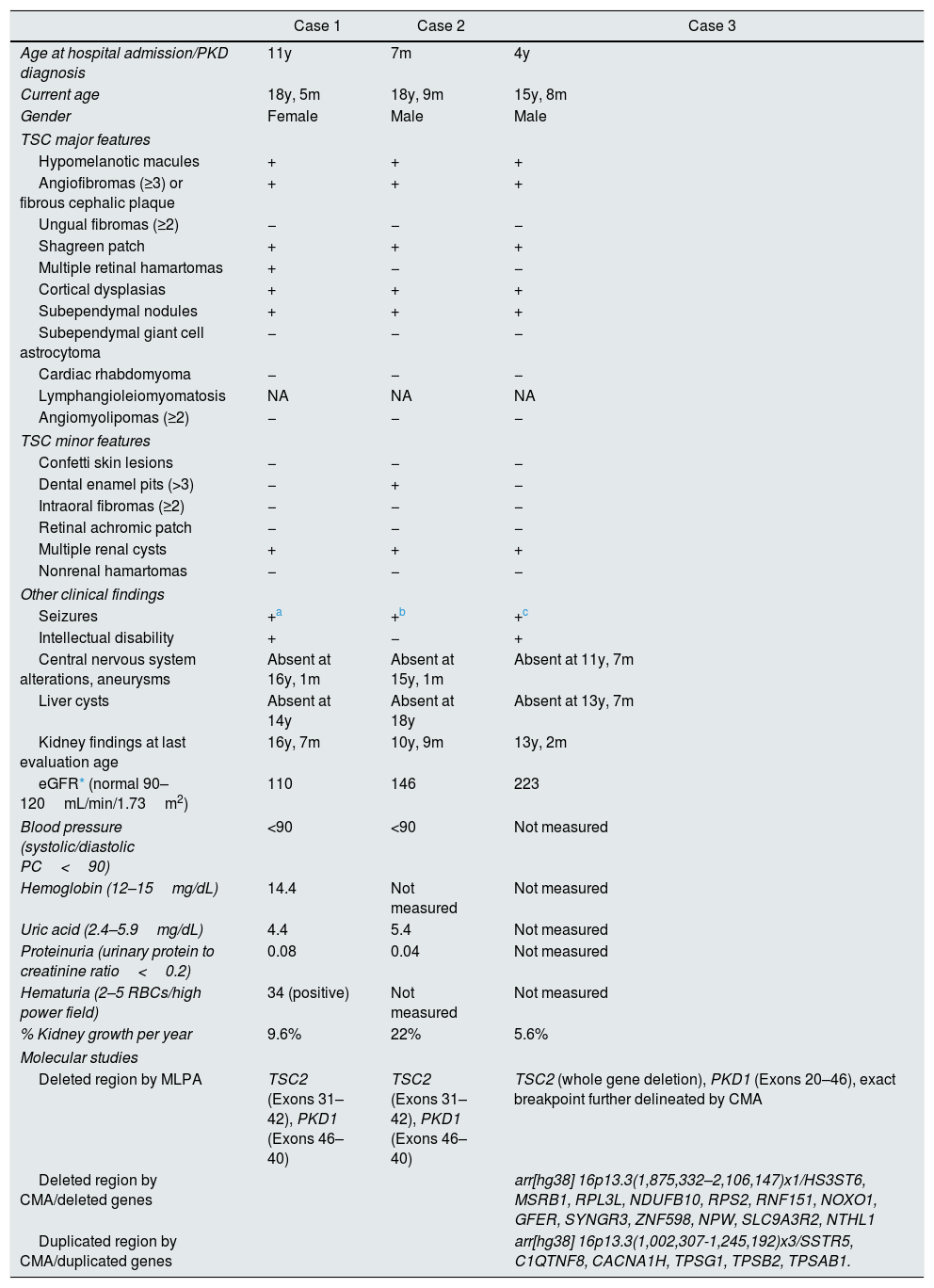
Herein we describe three unrelated Mexican pediatric patients who fulfilled the criteria of Northrup et al. for a definitive diagnosis of TSC (Table 1).17 Written informed consent was obtained for all cases and the five available parents, besides, the study protocol was revised and approved by the local ethics committee (reference number 060/2014). DNA samples were acquired from peripheral blood leukocytes by standard in silica adsorption method. At the time of admission, cases 1 and 2 revealed multiple bilateral renal cysts on ultrasonography (US) (Fig. 2A and B). Since this suggested a classic PKDTS phenotype, these cases were subjected to a multiplex ligation-dependent probe amplification (MLPA) assay (SALSA MLPA® P337 TSC2 probemix, Lot 0510-A2; MRC-Holland Amsterdam, The Netherlands) that included 39 probes for TSC2 and one probe for PKD1 (exon 40). The MLPA results revealed a deletion involving exons 31–42 from TSC2 and at least exons 40–46 from PKD1 (Table 1, Fig. 3A) in both cases.
Clinical features of the three studied cases of PKDTS.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Age at hospital admission/PKD diagnosis | 11y | 7m | 4y |
| Current age | 18y, 5m | 18y, 9m | 15y, 8m |
| Gender | Female | Male | Male |
| TSC major features | |||
| Hypomelanotic macules | + | + | + |
| Angiofibromas (≥3) or fibrous cephalic plaque | + | + | + |
| Ungual fibromas (≥2) | − | − | − |
| Shagreen patch | + | + | + |
| Multiple retinal hamartomas | + | − | − |
| Cortical dysplasias | + | + | + |
| Subependymal nodules | + | + | + |
| Subependymal giant cell astrocytoma | − | − | − |
| Cardiac rhabdomyoma | − | − | − |
| Lymphangioleiomyomatosis | NA | NA | NA |
| Angiomyolipomas (≥2) | − | − | − |
| TSC minor features | |||
| Confetti skin lesions | − | − | − |
| Dental enamel pits (>3) | − | + | − |
| Intraoral fibromas (≥2) | − | − | − |
| Retinal achromic patch | − | − | − |
| Multiple renal cysts | + | + | + |
| Nonrenal hamartomas | − | − | − |
| Other clinical findings | |||
| Seizures | +a | +b | +c |
| Intellectual disability | + | − | + |
| Central nervous system alterations, aneurysms | Absent at 16y, 1m | Absent at 15y, 1m | Absent at 11y, 7m |
| Liver cysts | Absent at 14y | Absent at 18y | Absent at 13y, 7m |
| Kidney findings at last evaluation age | 16y, 7m | 10y, 9m | 13y, 2m |
| eGFR* (normal 90–120mL/min/1.73m2) | 110 | 146 | 223 |
| Blood pressure (systolic/diastolic PC<90) | <90 | <90 | Not measured |
| Hemoglobin (12–15mg/dL) | 14.4 | Not measured | Not measured |
| Uric acid (2.4–5.9mg/dL) | 4.4 | 5.4 | Not measured |
| Proteinuria (urinary protein to creatinine ratio<0.2) | 0.08 | 0.04 | Not measured |
| Hematuria (2–5 RBCs/high power field) | 34 (positive) | Not measured | Not measured |
| % Kidney growth per year | 9.6% | 22% | 5.6% |
| Molecular studies | |||
| Deleted region by MLPA | TSC2 (Exons 31–42), PKD1 (Exons 46–40) | TSC2 (Exons 31–42), PKD1 (Exons 46–40) | TSC2 (whole gene deletion), PKD1 (Exons 20–46), exact breakpoint further delineated by CMA |
| Deleted region by CMA/deleted genes | arr[hg38] 16p13.3(1,875,332–2,106,147)x1/HS3ST6, MSRB1, RPL3L, NDUFB10, RPS2, RNF151, NOXO1, GFER, SYNGR3, ZNF598, NPW, SLC9A3R2, NTHL1 | ||
| Duplicated region by CMA/duplicated genes | arr[hg38] 16p13.3(1,002,307-1,245,192)x3/SSTR5, C1QTNF8, CACNA1H, TPSG1, TPSB2, TPSAB1. | ||
All cases are definitive according to the criteria of Northrup et al.17 Seizures are well controlled in all three cases.
CMA: chromosomal microarray; eGFR: estimated glomerular filtration rate; m: months; MLPA: multiple ligation probe amplification; NA: not analyzed; y: years; +: present; −: absent.
 Polycystic kidney disease detected by US in cases 1 and 2. (C) Single cyst in the right kidney detected by US in case 3 (arrows). (D) Polycystic kidney disease detected by MRI in case 3.")
 Schematic representation of the deletions detected by MLPA in the three PKDTS cases (not drawn to scale). Vertical gray lines in the TSC2 and PKD1 genes indicate exons with proportional spacing. The long black lines above indicate the range of each heterozygous deletion. A short dotted arrow and a question mark represent the expected minimal deletion interval estimated by MLPA. (B) Schematic representation of the rearrangement detected in the 16p13.3 region by CMA in case 3 (not drawn to scale). Gray lines at the right indicate deleted coding genes (complete deletion of TSC2 and deletion of exons 20–46 of PKD1) and double gray lines at the left indicate the duplicated coding genes (three copies), including SSTR5. Abbreviations: Cen: centromeric region, CMA: chromosomal microarray analysis, Ex: exon, MLPA: multiplex ligation-dependent probe amplification, Tel: telomeric region.")
Molecular PKDTS characterization in the three TSC patients. (A) Schematic representation of the deletions detected by MLPA in the three PKDTS cases (not drawn to scale). Vertical gray lines in the TSC2 and PKD1 genes indicate exons with proportional spacing. The long black lines above indicate the range of each heterozygous deletion. A short dotted arrow and a question mark represent the expected minimal deletion interval estimated by MLPA. (B) Schematic representation of the rearrangement detected in the 16p13.3 region by CMA in case 3 (not drawn to scale). Gray lines at the right indicate deleted coding genes (complete deletion of TSC2 and deletion of exons 20–46 of PKD1) and double gray lines at the left indicate the duplicated coding genes (three copies), including SSTR5. Abbreviations: Cen: centromeric region, CMA: chromosomal microarray analysis, Ex: exon, MLPA: multiplex ligation-dependent probe amplification, Tel: telomeric region.
For case 3, a single cyst (<10mm) in the right kidney was identified by US at 4 years of age, and follow-up US examinations yielded similar results (Fig. 2C). It should be noted that the molecular protocol carried out in this case 3 started with a direct Sanger sequencing (SS) of all coding exons and the exon–intron boundaries of TSC1 and TSC2 since there was no clinical suspicious of PKDTS due to the presence of a single renal cyst. As the SS results did not reveal any pathogenic variant a MLPA methodology was implemented and it revealed the presence of a large deletion that comprised all coding exons of TSC2 and at least exons 40–46 of PKD1 (Fig. 3A). Based on this genotype, a posteriori magnetic resonance imaging (MRI) was performed at age 15 to intentionally search for multiple renal cysts. The MRI results exposed more than 25 small cysts of variable size (9mm±SD 5.39mm) in both kidneys (Fig. 2D).
To clarify the size and exact breakpoints of the deleted region in case 3, we performed chromosomal microarray (CMA) analysis (CytoScan™ High Density Microarray; Affymetrix, Inc., Santa Clara, CA, USA) using the Human Genome Assembly hg38 for the analysis. We detected a 230.8kb heterozygous deletion that encompassed 13 coding genes, including all of TSC2 and part of PKD1 (exons 20–46), along with a neighboring heterozygous duplication of a previously undescribed CNV (242.9kb) in the 16p13.3 region that included six coding genes (Fig. 3B).
At the time of last evaluation, renal function was preserved in all three cases, although cases 2 and 3 had elevated glomerular filtration rates, and case 1 had microscopic hematuria without proteinuria (Table 1). None of the three cases presented intracranial aneurysm (Table 1). Both parents of all three patients had normal findings on clinical examination, cerebral computed tomography and renal US, while five had normal MLPA TSC2 results. Thus, only two out of the three PKDTS cases identified herein were considered as the novo since DNA was not available for testing in the father of case 2.
DiscussionThe three studied cases displayed multiple renal cysts. Cases 1 and 2 were consistent with a classic PKDTS phenotype: large kidney cysts were identified by US at an early stage and predominated over angiomyolipomas (Fig. 2A and B, Table 1). These two cases appeared to exhibit the same partial heterozygous deletion of TSC2 and PKD1, although the exact 5́ deletion breakpoint in PKD1 was not identified by the employed MLPA assay (Fig. 3A). To date, 30 PKDTS cases have been described in which the contiguous deletion affects parts of TSC2 and PKD1 (Fig. 1). In general, regardless of the extent of the deletion, the renal cysts were larger and the renal function fell below normal at a younger age than seen in typical TSC cases without any PKD1 deletion.10 Unexpectedly, case 3 exhibited small cysts that could only be detected by MRI, which is better able to detect small cysts (<1cm) in ADPKD patients compared to US, although the latter is the initial test of choice in the polycystic kidney disease.18
To date, at least 25 other PKDTS cases have been reported to include deletion of genes in addition to TSC2 and PKD1 (Fig. 1); among them, our case 3 appears to harbor the largest deletion upstream of TSC2 (Fig. 3B). In three of the previously reported cases, (Fig. 1) the early PKD symptoms and end-stage renal disease were recognized fairly late (>20 years).8,11,12 This is similar to case 3, in which only a few small cysts were found at age 15 and renal function was preserved (Table 1). The previous authors speculated that the relatively mild kidney involvement could reflect somatic mosaicism or the presence of genetic modifiers, but these hypotheses were not tested.8,11
Other studies have suggested that a mild PKD phenotype (better preserved renal function, as evaluated by glomerular filtration rate, and cystic disease recognized only in adult life) is associated with low/high grade deletion mosaicism.6,10,11 However, the apparently mild PKD phenotype in case 3 could not be explained by somatic mosaicism, as the MLPA results showed that the ratio values (STD±0.10) for each probe involved in the TSC2/PKD1 contiguous deletion were reduced by 0.5, implying that 100% of the peripheral leukocyte cells were heterozygous for this genotype and in addition, we detected a smooth signal of 1.2 copies (heterozygous deletion genotype) in CMA data. However, a low-level mosaicism could not be totally ruled out by CMA data since the deletion size is very small and the CytoScanTM algorithm is designed to discard mosaicism by the analysis of at least 5000 markers in deleted region.
The variable progression of PKD in our patients and the reported cases plus the absence of a clear genotype–phenotype correlation suggest that the mechanisms underlying renal cyst formation in PKD are extremely complex and may involve environmental and/or genetic factors that are not solely related to the PKD genotype. The identified 296.7kb CNV in the 16p13.3 region of case 3, which had not been previously reported as pathogenic in Decipher19 or the Database of Genomic Variants,20 included six coding genes and remarkable among them is the SSTR5 (somatostatin receptor) gene. Although, small clinical trials showed that somatostatin analogs (SSAs) decreased total kidney volume in patients with ADPKD, larger trials are ongoing to know their final effect in kidney cysts, so at this time, they are only indicated in ADPKD patients with severe liver cystic disease.21–25 The precise mechanism through which SSAs (e.g., lanreotide and octreotide) inhibit cystogenesis is not fully understood, but SSAs can inhibit cAMP accumulation, which plays a crucial role in cystogenesis.26,27 We hypothesize that the triple gene dosage of SSTR5 correlates with increased binding of the receptor's ligand, which would further reduce intracellular cAMP levels and decrease cellular proliferation through the Ras/Raf/MEK/ERK pathways.27 Modifier genes have previously been proposed to be present within CNVs of monogenic disorders. For example, Artuso et al.,28 detected a CNV containing a candidate modifier gene for Rett syndrome (MIM#312750). In two sisters with the same pathogenic genotype of MECP2 (the gene responsible for Rett syndrome), the three copies of CROCC (1p36.13) was associated with a milder phenotype, while a single gene copy was associated with the classic phenotype of Rett syndrome. Thus, it can be hypothesized that the presence of the CNV with the three SSTR5 copies in our case 3 may counteract the TSC2 and PKD1 deletion and modulate the PKD severity. However, future work is warranted to examine this possibility.
In the other hand, we can neither rule out that in case 3 the heterozygous loss of any of the 13 coding genes, others than TSC2 and PKD1, could act as disease modifier. In fact, it has been shown that some of these genes are expressed in the kidney,29 although none has been involved in a syndrome associating with kidney alterations in OMIM.30
ConclusionsThe cystic phenotype in PKDTS cases varies considerable among individuals; this suggests that a combination of factors besides the TSC2/PKD1 genotype, as modifying genes could be implied in the progression of the disease. Given the reported effect of SSAs in cystogenesis and the presence of three copies of SSTR5 in the patient who exhibited the mildest PKD phenotype (case 3) we hypothesize that an increased gene dosage of SSTR5 could counteract the PKD phenotype and its progression. The main limitation of our work relies on the fact that the exact 5′PKD1 breakpoint in cases 1 and 2 could not be determined, however, these two cases presented multiple cysts in both kidneys at early age as expected with a classic PKDTS. Considering that no previous studies have reported possible modifier genetic variants in other PKDTS patients, future work that includes functional analyses, frequency population studies in healthy individuals and more PKDTS cases will be necessary to determine the role of the 16p13.3 CNV, that contains SSTR5 in case 3 which apparently harbors the largest TSC2/PKD1 deletion and the mildest PKDTS phenotype.
FundingThis study was supported by the research funding from the National Institute of Pediatrics, Ciudad de México, México (Recursos Fiscales del Programa E022), as well as funding from Consejo Nacional de Ciencia y Tecnología, México (CONACyT FONSEC SSA/IMSS/ISSSTE, S0008, 2016–2018, Project #261404), Fundación “Miguel Alemán” 2012 and Novartis (2013).
Conflict of interestThe authors declare no conflicts of interests.
We thank Dr. Lorena Lechuga Becerra, MD, for participating in the initial medical assessment of the patients. This study makes use of data generated by the DECIPHER community. A full list of centers that contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from www.decipher.sanger.ac.uk.



