


La acidosis tubular renal distal (ATRD) es una enfermedad rara que se debe al fallo del proceso normal de acidificación de la orina a nivel tubular distal y colector. Se caracteriza por una acidosis metabólica hiperclorémica persistente, con anión gap normal en plasma, en presencia de un pH urinario elevado y baja excreción urinaria de amonio.
Se han descrito hasta el momento 5 genes cuyas mutaciones dan lugar a ATRD primaria. Las alteraciones de los genes ATP6V1B1 y ATP6V0A4 se heredan de forma recesiva y están asociadas a formas de inicio más precoces y con sordera neurosensorial en muchos casos. Las variantes patogénicas en el gen SLC4A1 se heredan habitualmente de forma dominante y dan lugar a cuadros más leves, con un diagnóstico más tardío y alteraciones electrolíticas menores. Sin embargo, la evolución a nefrocalcinosis y litiasis, y el desarrollo de enfermedad renal crónica a medio-largo plazo se ha descrito de forma similar en estos 3grupos. Por último, se han descrito también formas recesivas de ATRD asociadas a mutaciones en los genes FOXI1 y WDR72.
El manejo clínico de la ATRD se basa en sales de bicarbonato o citrato, que no logran corregir en todos los casos las alteraciones metabólicas descritas y, por lo tanto, las consecuencias asociadas a ellas. Recientemente, un nuevo tratamiento basado en sales de bicarbonato y citrato de liberación prolongada ha recibido la denominación de medicamento huérfano en Europa para el tratamiento de la ATRD.
Distal renal tubular acidosis (DRTA) is a rare disease resulting from a failure in the normal urine acidification process at the distal tubule and collecting duct level. It is characterised by persistent hyperchloremic metabolic acidosis, with a normal anion gap in plasma, in the presence of high urinary pH and low urinary excretion of ammonium.
To date, 5 genes whose mutations give rise to primary DRTA have been described. Alterations in the ATP6V1B1 and ATP6V0A4 genes are inherited recessively and are associated with forms of early onset and, in many cases, with neurosensorial deafness. Pathogenic variants in the SLC4A1 gene are habitually inherited dominantly and give rise to milder symptoms, with a later diagnosis and milder electrolytic alterations. Nonetheless, evolution to nephrocalcinosis and lithiasis, and the development of chronic kidney disease in the medium to long term has been described in a similar manner in all 3groups. Lastly, recessive forms of DTRA associated to mutations in the FOXI1 and WDR72 genes have also been described.
The clinical management of DTRA is based on bicarbonate or citrate salts, which do not succeed in correcting all cases of the metabolic alterations described and, thus, the consequences associated with them. Recently, a new treatment based on slow-release bicarbonate and citrate salts has received the designation of orphan drug in Europe for the treatment of DTRA.
- –
La ATRD hereditaria se debe a mutaciones en los genes que codifican los transportadores que regulan el equilibrio ácido base a nivel tubular distal.
- –
Las mutaciones en los genes ATP6V0A4 y ATV6V1B1 se asocian a formas más precoces de ATRD, con desarrollo frecuente de sordera neurosensorial desde la infancia.
- –
La evolución a enfermedad renal crónica es frecuente en todas las formas hereditarias y se debe a diversos factores, fundamentalmente la nefrocalcinosis/litiasis renal asociada, la hipopotasemia crónica y las pielonefritis agudas recurrentes. Además, un mal control metabólico puede empeorar el pronóstico de la función renal.
- –
El tratamiento tradicional con sales de bicarbonato o citrato no logra un óptimo control metabólico en todos los casos.
La acidosis tubular renal distal (ATRD) es una enfermedad poco común con una incidencia estimada < 1:100.0001. También conocida como ATR tipo 1 o ATR clásica, se caracteriza por una acidosis metabólica hiperclorémica persistente, con anión gap normal en plasma, en presencia de un pH urinario elevado y baja excreción urinaria de amonio. La ATRD se debe al fallo de las células intercaladas renales tipo A del túbulo colector para acidificar la orina de manera normal, consecuencia de una disfunción en cualquiera de los transportadores involucrados en este proceso1,2.
EtiologíaLa ATRD puede ser adquirida o heredada. Las formas adquiridas generalmente suelen ocurrir en adultos y pueden estar causadas por el uso de medicamentos (sobre todo antimicrobianos, antiinflamatorios, diuréticos y medicamentos antivirales)3, por enfermedades autoinmunes (síndrome de Sjögren, lupus eritematoso sistémico, colangitis biliar primaria, colangitis esclerosante primaria, hepatitis autoinmune y tiroiditis autoinmune)4,5 o bien pueden ser secundarias a uropatías o a un trasplante renal. Se desconocen con detalle los mecanismos que dan lugar a estas formas adquiridas, que pueden deberse a alteraciones voltaje dependientes, falta de diferencia transepitelial negativa en la luz distal o defectos de gradiente6.
En pacientes pediátricos, la etiología más frecuente es la hereditaria y se debe a alteraciones en los genes que codifican o controlan la codificación de los canales implicados en la acidificación urinaria a nivel del túbulo distal y colector (fig. 1)2,6,7. Actualmente, se reconocen 5genes cuyas mutaciones pueden dar lugar a ATRD: ATP6V0A4, ATP6V1B1, SLC4A1, FOXI1 y WDR7.

Transportadores ácido-base implicados en la secreción de H+ y reabsorción de HCO3– en las células alfa intercaladas del túbulo colector. La secreción de H+ está mediada por la H+-ATPasa vacuolar, que transfiere este protón activamente a través de la membrana luminal. El H+ puede también ser secretado gracias a una segunda ATPasa, la H+/K+-ATPasa que intercambia H+ por K. El bicarbonato, formado intracelularmente por la acción de la CAII, abandona la célula a través de la membrana basolateral, mediante la proteína trasportadora AE1 o proteína banda 3.
Los defectos en la actividad de la bomba H+ ATPasa (V-ATPasa) causan la mayoría de los casos primarios de ATRD. La H+-ATPasa es una bomba de protones altamente conservada que se expresa en las células alfa intercaladas y que está formada por 2dominios, el V1 y V0. La subunidad B1, codificada por el gen ATP6V1B1, forma parte del dominio V1 que captura protones del citoplasma celular. Esta subunidad se expresa además en las células del oído interno y del saco endolinfático, entre otras8,9. La subunidad A4, codificada por el gen ATP6V0A4, es parte del dominio transmembrana V0 involucrado en la translocación de protones a través de la membrana celular y se expresa en riñón, oído interno y epidídimo9,10.
Este tipo de ATRD sigue un patrón de herencia autosómico recesivo. Entre las mutaciones más frecuentes en estos genes se encuentran las mutaciones nonsense, frameshift o de splice-site, que se prevé que alteren la proteína codificada, mientras que solo se han descrito mutaciones missense en unos pocos pacientes8,11-13. Experimentos en modelos de cultivos celulares han demostrado que la mayoría de mutaciones identificadas en esta subunidad B1 causan una disfunción o una alteración en el ensamblaje del complejo proteico que forma la bomba V-ATPasa14. Las mutaciones en la subunidad A4 pueden afectar a la unión de estas 2subunidades, lo que conduce a un ensamblaje incorrecto de los dominios V1 y V0, formando una V-ATPasa estructural y funcionalmente defectuosa15.
En Europa, las mutaciones en los genes ATP6V1B1 y ATP6V0A4 parecen ser las más frecuentes16. En nuestra experiencia, la mayoría de los pacientes del norte de España presentan mutaciones en el gen ATP6V0A4. Esto concuerda con estudios previos que reportan una mayor frecuencia de mutaciones en este gen respecto al gen ATP6V1B1 en la población española y europea6,17. Entre ellas llama especialmente la atención la presencia de la variante c.1691+2dup en diferentes pacientes de nuestra cohorte, como reflejo de un posible efecto fundador en esta región geográfica (datos pendientes de publicación).
Mutaciones en el gen SLC4A1El gen SLC4A1 presenta un papel crucial en la homeostasis ácido-base ya que codifica un intercambiador de cloruro-bicarbonato, también conocido como AE1 o proteína banda 3, responsable de la reabsorción de HCO3− junto con la excreción de cloruro18. Esta proteína se expresa en la membrana plasmática de los eritrocitos y en la membrana basolateral de las células alfa intercaladas renales de los túbulos colectores19.
La herencia de este tipo de ATRD es compleja, con formas de trasmisión tanto autosómica dominante, sobre todo en caucásicos, como recesiva, más común en asiáticos20,21. Las manifestaciones clínicas son más severas en los pacientes con ATRD de herencia recesiva22.
Se han descrito diferentes tipos de mutaciones, que en estudios experimentales causan retención intracelular de la proteína mutada, reducción de su actividad de transporte, mal plegamiento y degradación o, incluso, erróneo direccionamiento hacia la membrana luminal23,24. La mutación de herencia recesiva más común, G701D, causa ATRD que se asocia con anemia hemolítica en algunos casos16. Por otro lado, entre los pacientes de origen caucásico la mutación de herencia autosómica dominante más común es R589H17. La introducción de esta mutación en ratones da lugar a la disfunción de las células intercaladas con una expresión reducida de las bombas de protones16,25. En ratones se ha observado que la ausencia completa del intercambiador AE1 provoca una acidosis metabólica severa, mientras que los heterocigotos no mostraban ningún defecto aparente16.
Se han descrito pocos casos de ATRD con defectos en este canal en nuestra población y tienen habitualmente un fenotipo más leve con un inicio tardío respecto a las mutaciones en los genes descritos previamente9,26,27.
Otros genes relacionados con acidosis tubular renal distalLos genes descritos previamente únicamente explican el 70-80% de los casos de ATRD primaria, lo cual respalda la existencia de otros genes candidatos responsables de un número significativo de casos17,28.
Recientemente se han descrito variantes patogénicas en homocigosis en el gen FOXI1 como responsables de sordera neurosensorial de aparición temprana y ATRD autosómico recesiva29. FOXI1 es un factor de transcripción crucial para la regulación de diversos transportadores de membrana, entre los que se incluyen aquellos necesarios para una correcta acidificación a nivel del túbulo distal y el oído interno (AE1, AE4 y varias subunidades de la V-ATPasa)30. Enerbäck et al.29 demostraron que en pacientes con mutaciones homocigotas con pérdida de función en este factor, hay una actividad de unión al ADN de FOXI1 muy limitada, lo que probablemente resulte en una incapacidad para la transactivación de algunas de estas proteínas transportadoras, lo cual provoca un síndrome grave de sordera y acidosis.
Asimismo, se han descrito recientemente variantes patogénicas en homocigosis en el gen WDR72 como causa de ATRD hereditaria31. Se cree que este gen podría estar implicado en el tráfico intracelular, afectando al direccionamiento de las proteínas reguladoras del equilibrio ácido-base, como la isoforma del transportador AE1 o la V-ATPasa, provocando la retención intracelular o direccionamiento erróneo de las mismas31. Las mutaciones en WDR72 se han descrito previamente asociadas a amelogénesis imperfecta, que incluye un grupo amplio de enfermedades hereditarias que afectan a la formación del esmalte dental32.
Diferentes estudios en modelos animales han revelado nuevos genes que podrían estar implicados en la ATRD. Algunos han sido identificados en ratones, pero no hay evidencia de que causen ATRD en humanos. Recientemente, estudios de secuenciación de exoma completo en pacientes con ATRD han revelado al gen ATP6V1C2 como un nuevo gen responsable de ATRD recesiva33. Este gen codifica la subunidad C de la V-ATPasa y se expresa mayormente en las células intercaladas del túbulo colector.
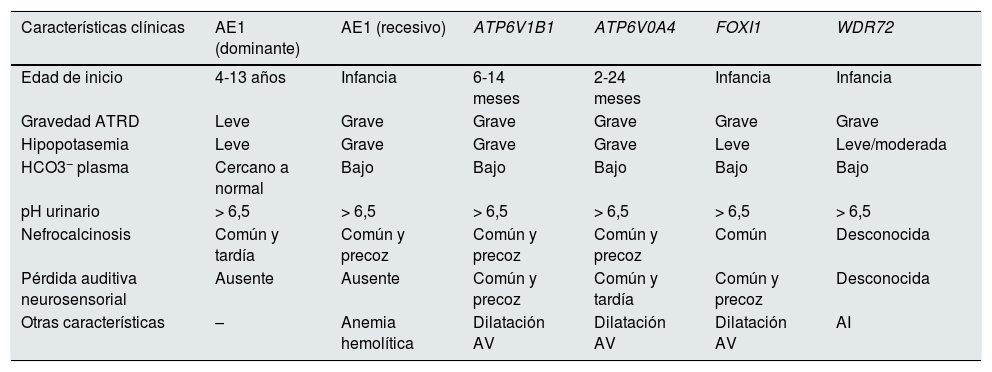
Manifestaciones clínicas y correlación genotipo-fenotipoLas manifestaciones clínicas de estos pacientes son comunes, aunque presentan alguna particularidad según el gen causal subyacente (tabla 1).
Diferencias en las características clínicas entre los grupos genéticos principales que causan ATRD
| Características clínicas | AE1 (dominante) | AE1 (recesivo) | ATP6V1B1 | ATP6V0A4 | FOXI1 | WDR72 |
|---|---|---|---|---|---|---|
| Edad de inicio | 4-13 años | Infancia | 6-14 meses | 2-24 meses | Infancia | Infancia |
| Gravedad ATRD | Leve | Grave | Grave | Grave | Grave | Grave |
| Hipopotasemia | Leve | Grave | Grave | Grave | Leve | Leve/moderada |
| HCO3− plasma | Cercano a normal | Bajo | Bajo | Bajo | Bajo | Bajo |
| pH urinario | > 6,5 | > 6,5 | > 6,5 | > 6,5 | > 6,5 | > 6,5 |
| Nefrocalcinosis | Común y tardía | Común y precoz | Común y precoz | Común y precoz | Común | Desconocida |
| Pérdida auditiva neurosensorial | Ausente | Ausente | Común y precoz | Común y tardía | Común y precoz | Desconocida |
| Otras características | – | Anemia hemolítica | Dilatación AV | Dilatación AV | Dilatación AV | AI |
AI: amelogénesis imperfecta; ATRD: acidosis tubular renal distal; dilatación AV: dilatación del acueducto vestibular.
Forma de presentación. Los pacientes con ATRD hereditaria inician habitualmente en la infancia con fallo de medro, vómitos de repetición, poliuria y episodios de descompensación aguda coincidiendo generalmente con cuadros infecciosos17,28. Los hallazgos a nivel bioquímico incluyen una acidosis metabólica marcada con anión gap normal por ausencia de otros ácidos que funcionen como aniones, y orina descompensada, con un pH generalmente mayor de 6. El anión gap urinario es positivo como reflejo de la escasa excreción de amonio. La hipopotasemia es frecuente en estos pacientes (30-50%), debido al hiperaldosteronismo secundario relativo y al defecto de gradiente en el túbulo distal, y produce debilidad muscular, estreñimiento, incapacidad para concentrar la orina y, en casos extremos, parálisis y arritmias cardiacas que pueden ser fatales6,34,35. En general, las personas con ATRD recesiva (ATP6V0A4 y ATP6V1B1) tienen acidosis metabólica e hipopotasemia más severa que aquellos con herencia dominante, que suelen presentar una forma más leve de acidosis parcialmente compensada36,37. La poliuria secundaria a un déficit de concentración urinaria es muy habitual en estos pacientes y, aunque los mecanismos causantes no han sido completamente estudiados, probablemente es causada por las anomalías bioquímicas asociadas, fundamentalmente la hipopotasemia y la hipercalciuria38. En algunos pacientes se observa al diagnóstico una disfunción asociada del túbulo proximal transitoria, que puede confundirse con un síndrome de Fanconi39,40. En ocasiones, pueden presentar asimismo hiperamonemia, que tiende a resolverse cuando se corrige la acidosis17,41.
En cuanto a la edad de inicio, los casos de ATRD recesivo (ATP6V0A4 y ATP6V1B1) se asocian con una presentación de los síntomas más temprana, generalmente entre los 6 y los 24 meses1,28. Sin embargo, los pacientes con una ATRD dominante (SLC4A1), tienen un inicio clínico más tardío, con una edad media al diagnóstico de entre 4 y 13 años1,9,28,36,37.
Crecimiento. La ATRD frecuentemente se diagnostica durante la evaluación de problemas de crecimiento en la infancia, principalmente con un crecimiento lineal deficiente con peso normal17. Esto se atribuye fundamentalmente al efecto inhibitorio de la acidosis metabólica en el metabolismo de la hormona de crecimiento. Los casos con mutaciones en el gen SLC4A1 tienden a presentar un retraso del crecimiento menos marcado que las formas recesivas debido a la presentación más tardía y la acidosis metabólica más leve36. Una terapia alcalina adecuada corrige el retraso de crecimiento, aunque algunas series recientes muestran que la talla final frecuentemente está por debajo de la media, independientemente del grupo genético1,17,42.
Hipoacusia. Debido a la importancia de la secreción de protones en la endolinfa para el funcionamiento del oído interno, la ATRD también se asocia a sordera neurosensorial. Esto ocurre en individuos con mutaciones en ATP6V0A4 y ATP6V1B1, debido a la expresión simultánea de la V-ATPasa en el riñón y oído, y en FOXI1. Sin embargo, la frecuencia de sordera asociada a mutaciones en ATP6V1B1 es mucho mayor, observándose en torno al 90% de los pacientes, respecto al 35-50% de los que tienen mutaciones en ATP6V0A41,28. Además, existe una diferencia significativa en la edad de aparición de la sordera entre estos 2grupos, siendo la detección de sordera en los primeros años de la vida altamente indicativa de una mutación subyacente en el gen ATP6V1B128,36. Algunos pacientes también presentan otras anomalías del sistema auditivo como, por ejemplo, la dilatación de los acueductos vestibulares, que, generalmente, es bilateral, y pueden sufrir mareos43,44. Los casos descritos con variantes patogénicas bialélicas en el gen FOXI1 presentan ATRD y sordera con dilatación del acueducto vestibular29.
Manifestaciones esqueléticas. La acidosis metabólica provoca la liberación de bicarbonato y fosfato del hueso, que actúan como amortiguadores alcalinos para restaurar el pH sanguíneo fisiológico. Esto da lugar a una desmineralización ósea que puede causar raquitismo en niños y osteomalacia en adultos. Estas anomalías se han descrito, sobre todo en el diagnóstico y de forma variable, en algunos pacientes con ATRD17,42,45.
Anemia hemolítica. Se han reportado algunos casos, mayoritariamente en el Sureste Asiático, en los que variantes patogénicas en el gen SLC4A1, de herencia recesiva, pueden dar lugar a ATRD y anemia hemolítica, generalmente en niños46. La importancia de este hecho recae en que estas variantes bialélicas pueden producir cambios morfológicos en los eritrocitos y estos, en condiciones de acidosis metabólica, son susceptibles de hemolizarse. La terapia alcalina corrige la anemia y la reticulocitosis. Estos pacientes también responden a transfusiones y terapia iónica42.
Manifestaciones renales. Los pacientes con ATRD desarrollan de forma muy frecuente y precoz una nefrocalcinosis o litiasis, debido a la combinación de hipercalciuria, hipocitraturia y un elevado pH urinario que favorece el depósito de cristales de oxalato y fosfato cálcico. La hipocitraturia es prácticamente universal en estos pacientes y se debe al aumento de reabsorción de citrato en el túbulo proximal como respuesta a la acidosis sistémica. La probabilidad de desarrollar nefrocalcinosis/litiasis se incrementa con la edad del paciente y con el retraso de inicio del tratamiento alcalinizante, es decir, con el diagnóstico tardío. La nefrocalcinosis se observa en el 90-95% de los pacientes, de cualquier grupo genético, y persiste en la mayoría de los casos a pesar de un adecuado control terapéutico. Una adecuada terapia alcalina suele evitar su progresión, pero no la revierte1,28,36. La frecuencia de litiasis en los pacientes con mutaciones en el SLC4A1 pudiera ser mayor debido posiblemente a un retraso en el diagnóstico causado por el fenotipo más leve que presentan1. Además, los pacientes con ATRD presentan con frecuencia pielonefritis agudas de repetición, asociadas a la presencia de litiasis potencialmente obstructivas y posiblemente a la hipercalciuria. Por último, muchos individuos desarrollan quistes medulares en la infancia o a edad adulta, que se ha atribuido en cierta medida a la hipopotasemia crónica. Sin embargo, otras tubulopatías con hipopotasemia mantenida, como el síndrome de Bartter, raramente presentan quistes medulares en la evolución17,47. Se desconoce su relevancia clínica ya que no se ha encontrado una correlación entre el desarrollo de quistes medulares y el grado de nefrocalcinosis o el deterioro de la función renal a largo plazo17.
Acidosis tubular renal distal incompletaSe han descrito casos de ATRD incompleta en pacientes portadores heterocigotos de variantes patogénicas en el gen ATP6V1B1. Estos presentan defectos leves de acidificación renal que no dan lugar a un pH sanguíneo alterado, pero no consiguen acidificar de forma adecuada la orina ante estímulo. En estos casos las pruebas funcionales para evaluar la capacidad máxima de acidificación urinaria pueden ser útiles. Estos individuos frecuentemente tienen una hipercalciuria e hipocitraturia asociada, y un riesgo elevado de cálculos renales42,48,49. De la misma manera, algunas mutaciones dominantes en el transportador AE1 pueden dar lugar a ATRD incompleta50.
EmbarazoLas mujeres con ATRD no presentan a priori trastornos de fertilidad asociados a su enfermedad. Sin embargo, durante el embarazo pueden presentar complicaciones metabólicas asociadas a la ATRD, fundamentalmente hipopotasemia y acidosis metabólica severas. Además, la hiperémesis gravídica puede dar lugar a descompensaciones hidroelectrolíticas al aumentar las pérdidas de electrólitos y dificultar la toma oral de suplementos. Otras complicaciones a tener en cuenta son las pielonefritis agudas recurrentes, ya de por sí habituales en los pacientes con ATRD, y la obstrucción ureteral por cálculos renales preexistentes. El empeoramiento de la función renal o la proteinuria no son complicaciones habitualmente observadas en las embarazadas con ATRD, salvo aquellos casos con enfermedad renal crónica (ERC) previa16,35.
Evolución a largo plazo de la función renalEn los últimos años, se han publicado algunas series amplias de pacientes con ATRD con diagnóstico molecular confirmatorio que evalúan el riesgo de ERC a largo plazo, describiéndose ERC grado ≥ 2 en un 30-80% de los pacientes, sin haber diferencias significativas entre los 3 genes principales implicados en la ATRD1,17,28. Los factores potencialmente implicados en esta progresión son el diagnóstico tardío, con un mayor número de episodios de descompensación y fallo renal agudo, la nefrocalcinosis o litiasis renal, particularmente las obstructivas, las pielonefritis de repetición, la presencia de quistes medulares y la hipopotasemia persistente21,28. Pese a que, como es esperable, el grado de ERC empeora con un mayor tiempo de evolución, muchos pacientes presentan ya ERC leve desde la adolescencia1,28.
A pesar de la tendencia a ERC descrita, la evolución clínica de los pacientes con ATRD es buena, en particular si el diagnóstico se establece de forma temprana con la consiguiente corrección de la acidosis. Aunque la ERC sea común, la enfermedad renal terminal es inusual. En muchos pacientes no se consigue una corrección adecuada de la acidosis y de los trastornos electrolíticos, lo cual refleja las dificultades con las formas de tratamiento con suplementos habituales, tanto en la tolerancia como en el mantenimiento de un cumplimiento terapéutico. Un adecuado control metabólico puede disminuir el riesgo de desarrollar ERC a largo plazo1.
Manejo de la acidosis tubular renal distal. Nuevos tratamientosEl objetivo principal en el manejo de la ATRD es la corrección de la acidosis metabólica y de las alteraciones secundarias y asociadas a ella, fundamentalmente la hipopotasemia, la hipercalciuria y la hipocitraturia. Con ello se pretende asegurar un crecimiento y un desarrollo normal en la infancia, evitar la desmineralización ósea y el raquitismo asociados, y evitar los factores de desarrollo de ERC a medio-largo plazo. El tratamiento alcalinizante, sin embargo, no logra modificar la aparición ni la evolución de la sordera neurosensorial.
La corrección de la acidosis se lleva a cabo con sales de bicarbonato o citrato, y la dosis habitualmente requerida para ello es mayor en el niño pequeño y va disminuyendo con el final del crecimiento. Así, en el lactante se precisan dosis de álcali de hasta 8 mEq/kg/día, posteriormente en el niño suele ser suficiente con dosis de 3 a 4 mEq/kg/día, y en el adulto generalmente no se precisan dosis mayores de 2-3 mEq/kg/día. El uso de citrato como alcalinizante tiene la ventaja de corregir la hipocitraturia asociada a la acidosis y de evitar en cierta medida el desarrollo de nefrocalcinosis asociada. El alcalinizante se usa asociado a sodio o potasio. El uso de sodio tiene sentido sobre todo en los pacientes con poliuria y depleción del volumen extravascular, pero tiene el inconveniente de aumentar la excreción de calcio, y de disminuir la reabsorción de bicarbonato a nivel proximal por la sobrecarga de volumen que puede conllevar. El uso de potasio intenta corregir además la hipopotasemia asociada.
El mayor inconveniente del uso de sales alcalinizantes, sobre todo citrato, es la mala tolerancia gastrointestinal. Además, es necesario dividir el tratamiento en varias dosis al día para mantener una homeostasis ácido-base constante. Esto hace que en muchas ocasiones no se logre un adecuado control metabólico1. Recientemente, se ha comercializado la molécula ADV7103, basada en citrato y bicarbonato potásico de liberación prolongada (2 administraciones diarias) con el fin de mejorar la adherencia terapéutica y el control metabólico en los pacientes con ATRD. Un estudio en fase 3ha demostrado su eficacia en la mejoría del control metabólico y su seguridad con una buena tolerancia gastrointestinal. Dicho tratamiento ha obtenido en Europa la designación de medicamento huérfano para el tratamiento de la ATRD.
Algunos grupos utilizan amilorida como diurético ahorrador de potasio para optimizar el tratamiento de la hipopotasemia y permitir reducir las dosis de suplemento de potasio2. En ocasiones se han usado las tiazidas como tratamiento de la hipercalciuria, con el fin de evitar la nefrocalcinosis y la progresión a ERC17. Sin embargo, este tratamiento no es recomendable de forma general al empeorar potencialmente la pérdida de potasio y no haber evidencias de que mejore la función renal a largo plazo.
Para evaluar las anomalías auditivas neurosensoriales, se debe realizar un audiograma estándar para explorar la conducción ósea y aérea enmascarada y no enmascarada a diferentes frecuencias. Se pueden utilizar tanto la resonancia magnética como la tomografía computarizada para diagnosticar la dilatación del acueducto vestibular16,43. Los dispositivos auditivos (audífonos o implantes cocleares) y la enseñanza de idiomas son primordiales para garantizar el desarrollo intelectual normal y la integración social de estos pacientes16.
FinanciaciónLa empresa farmacéutica Advicenne (Francia) ha proporcionado financiación para el estudio de pacientes con acidosis tubular renal distal, pero no ha participado en la realización de este artículo (beca BC/A/19/039).
Conflicto de interesesNo.



