




El conocimiento de las enfermedades podría equipararse al de la construcción de un edificio, de tal modo que el primer ladrillo es necesario para que se coloque después el segundo y, sobre este, un tercero, y así sucesivamente hasta llegar lo más cerca posible del final. En el caso de las tubulopatías, esa «construcción» fue harto dificultosa porque se requería una cumplida comprensión de la fisiología tubular renal, que se amplificó sobremanera cuando se desarrollaron las técnicas de biología molecular. Algunos peldaños de la historia del conocimiento de la hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (HFHN) se han colocado y escrito en España. Esta es la historia.
SOBRE LA DESCRIPCIÓN DE LA ENFERMEDAD Y ALGUNOS DE LOS NOMBRES CON LOS QUE SE NOMINÓ CUANDO AÚN NO SE SABÍA SI SE TRATABA DE UNA ENTIDAD INDIVIDUALIZADA. LOS PRIMEROS CASOS PUBLICADOS EN ESPAÑA
Aunque no se citan anomalías oculares, es posible que tres trabajos aparecidos a finales de los sesenta y en los años setenta contuvieran los primeros casos publicados de HFHN1-3. En 1969, Alfrey y Jenkins escribieron el resumen clínico de un varón de 16 años que padecía tetania desde los cinco1. La magnesemia era muy reducida (0,8 mg/dl), y el aclaramiento de magnesio, elevado para el grado de magnesemia (10 ml/min). El joven tenía nefrocalcinosis radiológica y poliuria demostrada por un defecto de concentración resistente a la pitresina (260 mOsm/l), lo que parecía contribuir a la hipopotasemia existente (2,5-3,2 mEq/l), dado el hiperaldosteronismo secundario. No había hipercalciuria, pero el paciente mostraba datos compatibles con insuficiencia renal (79 ml/min). En 1972, Michelis et al. publicaron un artículo basado en los datos de dos hermanos con nefrocalcinosis, poliuria resistente a vasopresina e hipomagnesemia (1,1-1,2 mEq/l) secundaria a una pérdida tubular; uno de ellos padecía convulsiones2. La bicarbonatemia del primero de los hermanos era reducida (19 mEq/l) con una cloremia ligeramente elevada (112 mEq/l), pero los valores del otro hermano (22 mEq/l y 105 mEq/l, respectivamente) eran normales. En la prueba de acidificación con cloruro amónico el pH urinario se redujo a 5,5 y 5,4, respectivamente, por lo que, sensu stricto, solo podían tener una acidosis tubular renal distal incompleta2. Es posible que el primero de los hermanos tuviera una pérdida proximal de bicarbonato. En todo caso, el tratamiento con sales alcalinas no influyó en los niveles de magnesemia. En el tercero de los artículos primigenios, se cita que los hermanos afectos tenían una acidosis tubular incompleta e hipocitraturia, insuficiencia renal moderada y datos similares a los ya citados3. Es llamativo que algunos de esos primeros pacientes padecieran pielonefritis agudas2,3.
En 1979, apareció el primer artículo en el que se relacionó la hipercalciuria con el coloboma macular bilateral4. Sus autores creyeron que estaban ante una nueva variante del síndrome óculo-renal, por tener algunas concordancias con el síndrome de Lowe. Uno de los dos hermanos, pertenecientes a una familia consanguínea, tenía asimismo una pérdida moderada tubular proximal de bicarbonato. Al coloboma se añadía una degeneración tapeto-retiniana.
En la Reunión Nacional de Nefrología Pediátrica correspondiente a ese mismo año (Oviedo, 1979), el Grupo de Zaragoza presentó los dos primeros casos españoles de «Hipercalciuria idiopática renal. Asociación con coloboma macular»5. Se sospechó que la hipercalciuria era de origen renal porque el cociente calcio/creatinina no se modificó tras una sobrecarga oral de calcio. Los niños, además del coloboma, tenían miopía y nistagmo.
En los años ochenta y hasta mediados de los noventa se publicaron bastantes artículos de la entidad firmados en España, lo que es digno de destacar teniendo en cuenta la rareza del cuadro que nos ocupa. Estos trabajos correspondían tanto a niños como a adultos6-12. La cosa no queda aquí, porque en 1982 Gil-Gibernau, miembro de la Sección de Oftalmología Pediátrica del Hospital de Vall d'Hebron de Barcelona, firmó como primer autor un artículo basado en cuatro casos de hipercalciuria idiopática infantil asociada a miopía bilateral congénita y coloboma macular atípico. No se determinó la magnesemia de los pacientes, que, por otra parte, tenían nefrocalcinosis y defecto de la capacidad de concentración13. El manuscrito tuvo trascendencia, porque en la segunda edición del reconocido libro de nefrología pediátrica editado por Edelmann Jr. se citaba el síndrome de Gil-Gibernau como un nuevo cuadro que asociaba hipercalciuria, miopía y coloboma macular14.
LA ENFERMEDAD SE INDIVIDUALIZA COMO UNA TUBULOPATÍA DISTINTA Y SE LE OTORGA SU NOMBRE DEFINITIVO. TODO ELLO, POR PARTE DE AUTORES ESPAÑOLES
La hipomagnesemia de origen tubular renal estaba sin sistematizar bien entrados los años ochenta. Juan Rodríguez Soriano et al., en 1987, en un artículo antológico describieron que, al menos, debían considerarse tres causas hereditarias de hipomagnesemia causadas por sendos defectos en la reabsorción tubular renal de magnesio, a saber, la hipomagnesemia aislada familiar, la hipomagnesemia-hipopotasemia familiar o síndrome de Gitelman que se confundía con frecuencia con el síndrome de Bartter y la hipomagnesemia-hipercalciuria familiar, de la que se habían descrito al menos 15 pacientes, a los que los autores añadieron tres nuevos casos15. Sobre esta última entidad, los autores escribieron que «la hipomagnesemia se acompañaba siempre de hipercalciuria y nefrocalcinosis. Las anomalías oculares tales como la miopía y el nistagmo horizontal están, a menudo, presentes. La hipermagnesiuria es de una mayor relevancia que en las entidades previas y refleja un Tm de reabsorción de magnesio reducido. El defecto debe estar situado en la rama ascendente del asa de Henle e implicar el transporte tanto de calcio como de magnesio». En efecto, poco después se demostraría que el defecto está ubicado donde habían intuido Rodríguez Soriano et al. Unos años después, los mismos autores estudiaron el defecto de acidificación existente en esta tubulopatía16. Las pruebas destinadas a estimular la capacidad de acidificación distal mostraron un descenso del pH urinario (4,7-5,6), pero la excreción de amonio estaba reducida (26-46 µmol/min por 1,73 m2, 35-54 µmol/100 ml tasa de filtrado glomerular). Además, la pCO2 urinaria no se elevó adecuadamente después de una sobrecarga de bicarbonato sódico (gradiente de pCO2 orina/sangre: 1,3-17,7 mmHg). Los autores argumentaron que este trastorno en la capacidad de acidificación distal podía explicarse por un defecto tanto en la transferencia tubular de amoníaco como en la secreción de hidrogeniones en el ducto colector medular, causado probablemente por una nefropatía intersticial medular.
En 1995, Manuel Praga et al. publicaron los datos clínicos y bioquímicos correspondientes a ocho pacientes pertenecientes a cinco familias. Los autores comprobaron que en esta tubulopatía, de herencia autosómica recesiva, existe un deterioro progresivo de la función glomerular renal y que el daño tubular no recidiva tras el trasplante12. El título de su artículo es el que ha pasado a la posteridad dando el nombre definitivo a esta enfermedad, es decir, «Hipomagnesemia familiar con hipercalciuria y nefrocalcinosis».
SE DESCUBRE QUE LA HIPOMAGNESEMIA FAMILIAR CON HIPERCALCIURIA Y NEFROCALCINOSIS ES LA PRIMERA TUBULOPATÍA CAUSADA POR LA DISFUNCIÓN DE UNAS PROTEÍNAS QUE SE EXPRESAN EN EL ESPACIO PARACELULAR DE LA RAMA ASCENDENTE DEL ASA DE HENLE
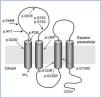
Desde principios de los setenta se sabía, mediante estudios de micropunción, que el magnesio se reabsorbe de forma predominante en la rama ascendente del asa de Henle17,18. En 1982, Shareghi y Agus demostraron en ese segmento tubular que la presencia de un voltaje transepitelial positivo es una fuerza importante que favorece la reabsorción neta de magnesio, calcio y potasio19 (figura 1). Asimismo, mediante estudios de micropunción, se supo que cantidades importantes de magnesio (50-60 %) se reabsorben en la rama ascendente gruesa del asa de Henle en comparación con las de sodio (20-25 %) o calcio (30-35 %)20. A principios de los noventa se disponía de la suficiente información para saber que el magnesio se reabsorbe por la vía paracelular20,21 (figura 1). Solo faltaba conocer si esa reabsorción estaba favorecida por la acción de determinadas proteínas. En efecto, en 1999, Simon et al. describieron la existencia de una proteína, la paracelina-1, que es necesaria para la reabsorción tubular paracelular de magnesio22. Esta proteína está localizada en las zonas de unión estrechas (tight junction), es decir, las estructuras intercelulares que permiten un contacto íntimo entre las células epiteliales adyacentes. En el mismo estudio firmado por Simon et al., se estableció que mutaciones en el gen PCLN-1 que codifica la paracelina-1 eran las causantes de la HFHN22,23. Al comprobarse que la paracelina-1 es un miembro de la familia de las claudinas, pasó a denominarse claudina-16 (gen CLDN16). Digamos, de paso, que el nombre de claudina es un homenaje a la doctora Philippa Claude, que en los años setenta estudió, mediante la ayuda del microscopio electrónico, las estructuras intercelulares conocidas como «zonula occludens»24.
Cuando ya se creía que se conocía todo sobre la enfermedad, surgieron dos hechos que precisaban una cumplida explicación. En primer lugar, la descripción realizada por Loris et al., basada en su serie constituida por nueve casos, en la que se citaba que, en los pacientes españoles, la frecuencia de alteraciones oculares era inusitadamente elevada (81 % frente a 24 % en pacientes procedentes de otros países)25. En segundo lugar, la observación de que existían pacientes con HFHN que no tenían mutaciones en el gen CLDN16. En este sentido, fue llamativo el hecho de que los pacientes españoles con HFHN, excepto muy pocos22,26, no tuvieran mutaciones en dicho gen27.
En 2006, Konrad et al. resolvieron la cuestión al identificar que los pacientes portadores de mutaciones en el gen CLDN19, otro miembro de la familia multigénica de las claudinas, padecían hipomagnesemia, enfermedad renal crónica (ERC) y anomalías oculares graves28. El producto del gen, la claudina-19, realiza su función, como la claudina-16, en las uniones estrechas del túbulo renal y la retina.
Un año después, se comprobó que, en condiciones fisiológicas, la claudina-19 actúa como una barrera selectiva a los cationes en las tight junctions y regula la permeabilidad a los cationes monovalentes y divalentes29. En 2008, Hou et al. demostraron que la claudina-16 interactúa con la claudina-1930, de tal modo que esta asociación confiere a las uniones estrechas la capacidad de contener un mecanismo selectivo en la reabsorción de cationes31.
LA MAYORÍA DE LOS PACIENTES ESPAÑOLES CON HIPOMAGNESEMIA FAMILIAR CON HIPERCALCIURIA Y NEFROCALCINOSIS SON PORTADORES DE LA MISMA MUTACIÓN EN EL GEN CLDN19. EL EFECTO FUNDADOR
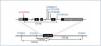
Hasta la fecha solo se han descrito unas pocas mutaciones en el gen CLDN1928,32,33 (figura 2). El estudio original de Konrad et al. incluía a siete pacientes españoles pertenecientes a distintas familias a los que se detectó una misma mutación en homocigosis, p.G20D (c.59G>A), en el gen CLDN19, a la que denominaron mutación española/hispánica28. La mutación da lugar al cambio de glicina por ácido aspártico en la posición 20 de la claudina-19 que altera el péptido señal y hace que la proteína mutante quede retenida dentro de la célula y no llegue a su localización habitual, que es la membrana celular, donde se localiza fisiológicamente la proteína normal28,30. Konrad et al. sugirieron que p.G20D es una mutación fundadora, es decir, que se originó en un antepasado común. La otra alternativa es que la mutación apareciera en un sitio propenso a mutarse (hot spot), en cuyo caso los individuos con la mutación pueden no estar relacionados y la secuencia del ADN alrededor de esta varía. Por el contrario, cuando se trata de una mutación fundadora, está ubicada en una región del cromosoma idéntica a la del «sujeto fundador». Vargas-Poussou et al. publicaron un estudio en el que describieron otros dos pacientes españoles no relacionados y 13 franceses, pertenecientes a 12 familias, con la mutación p.G20D, la mayoría en homocigosis32. Las familias que provenían del suroeste de Francia eran de origen español. Los datos obtenidos mediante un análisis de microsatélites en estas familias son consistentes con el efecto fundador32. Posteriormente, nuestro grupo ha publicado los resultados del estudio de una cohorte española compuesta por 34 pacientes con HFHN, 20 niñas y 14 niños, miembros de 30 familias aparentemente no relacionadas33,34. Los datos clínicos de estos pacientes muestran un alto riesgo de progresión hacia la ERC terminal (62 % con deterioro de la función renal y 20 % trasplantados), que concuerda con lo observado en la serie de los pacientes franceses32. Nuestros resultados también están de acuerdo con los del grupo francés en el alto porcentaje, 88 % y 91 %, respectivamente, de pacientes que presentan anomalías oculares severas como miopía magna, nistagmo y coloboma. En contraste, los nueve pacientes con mutaciones en el gen CLDN16 estudiados por el grupo francés, la mayoría de los cuales proviene del norte de África, no tienen anomalías oculares. El análisis de mutaciones en nuestra cohorte mostró que todos los pacientes tienen mutaciones en el gen CLDN19. La mutación p.G20D se detectó en 93 % de los pacientes no relacionados y estaba presente en homocigosis en el 83 % de los casos33,34. Ninguno de nuestros pacientes mostró mutaciones en CLDN16. De hecho, como se ha indicado más arriba, solo conocemos la existencia de tres pacientes españoles con una mutación en ese gen22,26. En nuestro estudio, determinamos los haplotipos de pacientes y familiares genotipando los tres microsatélites (D1S463, D1S193, D1S447) que flanquean el locus CLDN19 utilizados previamente en la cohorte francesa32 y cuatro polimorfismos de un solo nucleótido (SNPs, rs10890211, rs11210708, rs7548008, rs12141833) situados en los intrones 3 y 4 del gen33,34 (figura 3). Los resultados de estos análisis mostraron que los alelos con la mutación p.G20D exhiben un haplotipo común que se hereda con esta. Nuestros resultados, junto con los resultados previos de otros pacientes españoles o de origen español, indican la existencia de un efecto fundador responsable de la HFHN en España. La región alrededor de la mutación p.G20D compartida por los distintos pacientes es relativamente grande (unos 500 a 1000 kb), por lo que podemos suponer que la mutación fundadora se originó recientemente, quizás hace unos cientos de años, en un antecesor común a todas estas familias. El hecho de que la mayoría de nuestros pacientes presente la misma mutación en el gen CLDN19 facilita el diagnóstico molecular de la enfermedad en nuestro país. El análisis de la mutación p.G20D debe ser la primera exploración genética en pacientes con HFHN.
La patogénesis de la ERC en pacientes con HFHN es una incógnita. No se ha podido establecer una correlación entre el grado de la nefrocalcinosis y la progresión hacia la insuficiencia renal crónica. Sin embargo, se ha sugerido que el efecto de las mutaciones en CLDN16 en la función de claudina-16 puede ocurrir durante el desarrollo del riñón y que, por tanto, la temprana progresión hacia la ERC puede ser debida a un desarrollo anormal del riñón seguido por fibrosis y nefrocalcinosis32,35. Los resultados de un estudio realizado en un grupo de 71 pacientes con mutaciones en CLDN16 mostraron que los pacientes con mutaciones que dan lugar a una pérdida completa de función de la claudina-16 en ambos alelos presentan los síntomas de la enfermedad a una edad más temprana que los que tienen, al menos, un alelo mutante con función parcial; además, los pacientes que tienen mutaciones con pérdida completa de función progresan más rápidamente hacia la ERC35. Es decir, la función residual de algunos mutantes de claudina-16 retrasa la progresión hacia la ERC. En comparación con CLDN16, en el que se han identificado 40 mutaciones distintas, solo se han descrito 13 mutaciones en CLDN19. Todavía, no se han publicado correlaciones genotipo-fenotipo en pacientes con mutaciones en este último gen. Resultados recientes sugieren que los pacientes con HFHN que presentan mutaciones en CLDN19 tienen un mayor riesgo de desarrollar ERC que los pacientes con mutaciones en CLDN1632. Sin embargo, estos resultados hay que tratarlos con cautela, ya que la mayoría de los primeros son portadores de la mutación p.G20D que supone una pérdida completa de función.
En relación con las manifestaciones extrarrenales, los pacientes con mutaciones en CLDN19 presentan, en la mayoría de los casos, anomalías oculares severas que no se observan en pacientes con mutaciones en CLDN1628,32,35. Esta diferencia se podría explicar porque la claudina-19 se expresa, también, de manera prominente en el epitelio pigmentario de la retina fetal, mientras que la expresión de claudina-16 en dicho epitelio es muy baja28,36. Por otra parte, en unos pocos pacientes con mutaciones en CLDN19 se han descrito manifestaciones neuromusculares32,34. Esto se ha relacionado con las anomalías en el comportamiento observadas en un modelo de ratón deficiente en claudina-1937. La claudina-19 también se expresa en las uniones celulares de las células de Schwann, por lo que mutaciones en el gen que la codifica podrían dar lugar a deficiencias en el sistema nervioso periférico37.
AGRADECIMIENTOS
La investigación de nuestro grupo ha sido financiada por el Fondo de Investigación Sanitaria (Proyectos PI09/91009 y PI11/00342, cofinanciados por el Fondo Europeo de Desarrollo Regional). Los autores son miembros del grupo RenalTube (www.renaltube.com).
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.

Figura 1. Representación esquemática del transporte de iones en la rama ascendente gruesa del asa de Henle

Figura 2. Mutaciones descritas en el gen CLDN19(28,32,33) y su localización en el modelo de claudina-19.

Figura 3. Región del cromosoma 1p34.2 que contiene el locus CLDN19.



