





Presentamos el caso de un varón de 32 años, con síndrome de genes contiguos TSC2/PKD1, que le ocasiona esclerosis tuberosa (ET) y poliquistosis renal autosómica dominante simultáneamente. Evolucionó a enfermedad renal terminal y se realizó trasplante renal a los 12 años. Los riñones presentaban angiomiolipomas (AML), que son tumores benignos frecuentes en pacientes con ET. A los 17 años postrasplante, presentó un cuadro de dolor abdominal, anemización y hematoma retroperitoneal. Dicho hematoma se produjo por el sangrado de los AML. Como tratamiento se realizó embolización selectiva. Nuestro paciente podría haberse beneficiado en el momento del trasplante renal del tratamiento con inhibidores de mTOR. Este fármaco actúa como inmunosupresor y reductor tumoral en la ET, al disminuir el riesgo de rotura y hemorragia. En este paciente no se administró porque cuando se trasplantó no se conocía la relación de los inhibidores de mTOR con la ET. Este caso confirma que, a pesar de tratarse de pacientes trasplantados o en diálisis, el riesgo de sangrado por los AML persiste, por lo cual se propone realizar controles periódicos de los riñones propios y valorar la nefrectomía.
We report the case of a 32-year-old male diagnosed with TSC2/PKD1 contiguous gene syndrome, presenting with tuberous sclerosis (TS) and autosomal dominant polycystic kidney disease simultaneously. He progressed to end-stage renal disease and received a kidney transplant at the age of 12. The native kidneys presented angiomyolipomas (AML), which are common benign tumours in patients with TS. Seventeen years after transplantation, he presented with abdominal pain, anaemia and a retroperitoneal haematoma, the latter caused by renal AML bleeding. Selective embolisation was performed. Our patient could have benefited from the administration of mTOR inhibitors at transplant. This therapy is immunosuppressive and reduces the size of benign tumours in TS as well as the risk of rupture and bleeding. This patient did not receive mTOR inhibitors at the time of the transplant because the relationship between mTOR inhibitors and TS was unknown at that time. This case confirms the persistent risk of renal AML bleeding for both transplanted patients and patients on dialysis. As a result, we would recommend routine check-ups of native kidneys and nephrectomy assessment.
La poliquistosis renal autosómica dominante (PQRAD) es el trastorno renal hereditario más frecuente, con una incidencia poblacional entre 1/400-1.0001. Ocasiona el 10% de los casos de enfermedad renal crónica terminal (ERCT) en nuestro país2. La PQRAD es una enfermedad genéticamente heterogénea, 2 de cuyos genes causales son PKD1 y PKD23,4. El gen PKD1 se encuentra situado en el cromosoma 16p13, adyacente a TSC2, uno de los genes causantes de esclerosis tuberosa (ET)5,6. Esta enfermedad es causada por mutaciones en dicho gen o por mutaciones en el gen TSC1. Cuando se produce una deleción que implica a los genes PKD1 y TSC2, a causa de su proximidad cromosómica, estamos ante la presencia de un síndrome de genes contiguos (SGC)7. El síndrome se caracterizada por una clínica grave tanto de PQRAD como de ET. La presencia de riñones con nefromegalia quística al nacimiento es similar a la que se observa en estadios avanzados de PQRAD, que generalmente conducen a ERCT en la segunda o tercera décadas de la vida. Solo existe un caso reportado de un paciente con ambas enfermedades causadas por mutaciones independientes del gen PKD1 y del TSC28.
El gen TSC1 codifica para la proteína hamartina y el gen TSC2 para la proteína tuberina formando el complejo hamartina/tuberina9. Este complejo interviene y regula la señalización de la cascada mammalian target of rapamycin (mTOR) a través de la vía (Rheb/mTOR/p70S6K) controlando el crecimiento celular, la progresión del ciclo celular y la apoptosis. Las mutaciones en este complejo producen una desregulación de la cascada de mTOR que causa aumento de la proliferación celular. Adicionalmente, la poliquistina 1, proteína codificada por el gen PKD1, interactúa con el complejo hamartina/tuberina y suprime la actividad de mTOR10.
La ET es un trastorno autosómico dominante sistémico, con elevada penetrancia y una prevalencia de 1/6.00011. Clínicamente se manifiesta por lesiones cutáneas (angiofibromas, fibromas ungueales, manchas hipomelánicas), lesiones renales benignas llamadas angiomiolipomas (AML), linfangioleiomiomatosis pulmonar, lesiones cardíacas como rabdomiomas, lesiones neurológicas como tubers y astrocitomas. Su presentación clínica es muy variable: oscila desde mínimos o inapreciables síntomas de la enfermedad hasta una afectación neurológica severa11.
Los AML renales asociados con ET suelen ser múltiples y bilaterales, con una incidencia del 55-75% en función de la edad12. Están formados por células inmaduras de músculo liso, tejido graso y vascular. El principal peligro que conllevan es su riesgo de rotura y hemorragia, que puede llegar a provocar una situación crítica vital. Los AML se diagnostican con ecografía, pero lo más utilizado es la tomografía computarizada (TC), que permite delimitar mejor las lesiones. Cuando el diagnóstico es dificultoso, por el escaso componente graso, se recurre a la resonancia magnética.
Clásicamente las opciones terapéuticas para el tratamiento de los AML han sido la embolización, las nefrectomías parciales electivas o las nefrectomías urgentes para los AML sangrantes. La embolización puede ocasionar el conocido síndrome postembolización13,14. Consiste en una respuesta inflamatoria debido a la presencia de tejido necrótico secundario a la isquemia por la embolización. Las técnicas intervencionistas reducen la masa renal funcionante, lo cual en esta enfermedad crónica y multifocal resulta tremendamente deletéreo.
Los inhibidores de mTOR se han aprobado como terapia de primera línea para los AML en la ET15 y han mostrado cierta eficacia en la disminución de la progresión del volumen renal de la PQRAD, pero no así en el deterioro de función renal característico de ella16,17.
Los riñones poliquísticos suelen experimentar una reducción de volumen y un relativo escaso número de complicaciones tras el trasplante renal18. De la misma manera, se supone que la progresiva disminución del flujo renal debería ocasionar una disminución del tamaño de los AML o, al menos, una disminución del riesgo del sangrado, aunque dicha afirmación no deja de ser una hipótesis no contrastada.
El caso que se presenta muestra que dicha hipótesis no parece cierta y que el riesgo de sangrado perdura tras muchos años de terapia renal sustitutiva.
Presentamos el caso de un varón de 32 años, con antecedentes de SGC por deleción de TSC2/PKD1, diagnosticado a los 8 meses de vida en el contexto de retraso neurocognitivo. El estudio genético se realizó mediante análisis de deleciones/duplicaciones de uno o varios exones mediante la técnica multiplex-ligation-dependent probe amplification. El paciente era portador de una deleción de novo causante del SGC. Esta deleción elimina los exones 43 al 46 del gen PKD1 y los exones 37 al 42 del gen TSC2 (fig. 1). El paciente es un caso esporádico, es decir, que ni el padre ni la madre le han transmitido estas enfermedades. Clínicamente presentaba episodios de crisis comiciales parciales-generalizadas debido a la presencia de tubers corticales, así como retraso mental grave. Asimismo, se detectaron al nacer masas abdominales bilaterales que se identificaron como riñones poliquísticos. A los 11 años se observó deterioro de la función renal progresivo y precisó tratamiento renal sustitutivo 4 años más tarde, mediante programa de diálisis peritoneal. Al año siguiente, se trasplantó de donante renal cadáver y el tratamiento inmunosupresor fue con prednisona, ciclosporina y azatioprina. A los 26 años se le realizó una exéresis de un tumor de Köenen en el pie derecho. Presentó varios episodios de neumonías de repetición adquiridas en la comunidad.
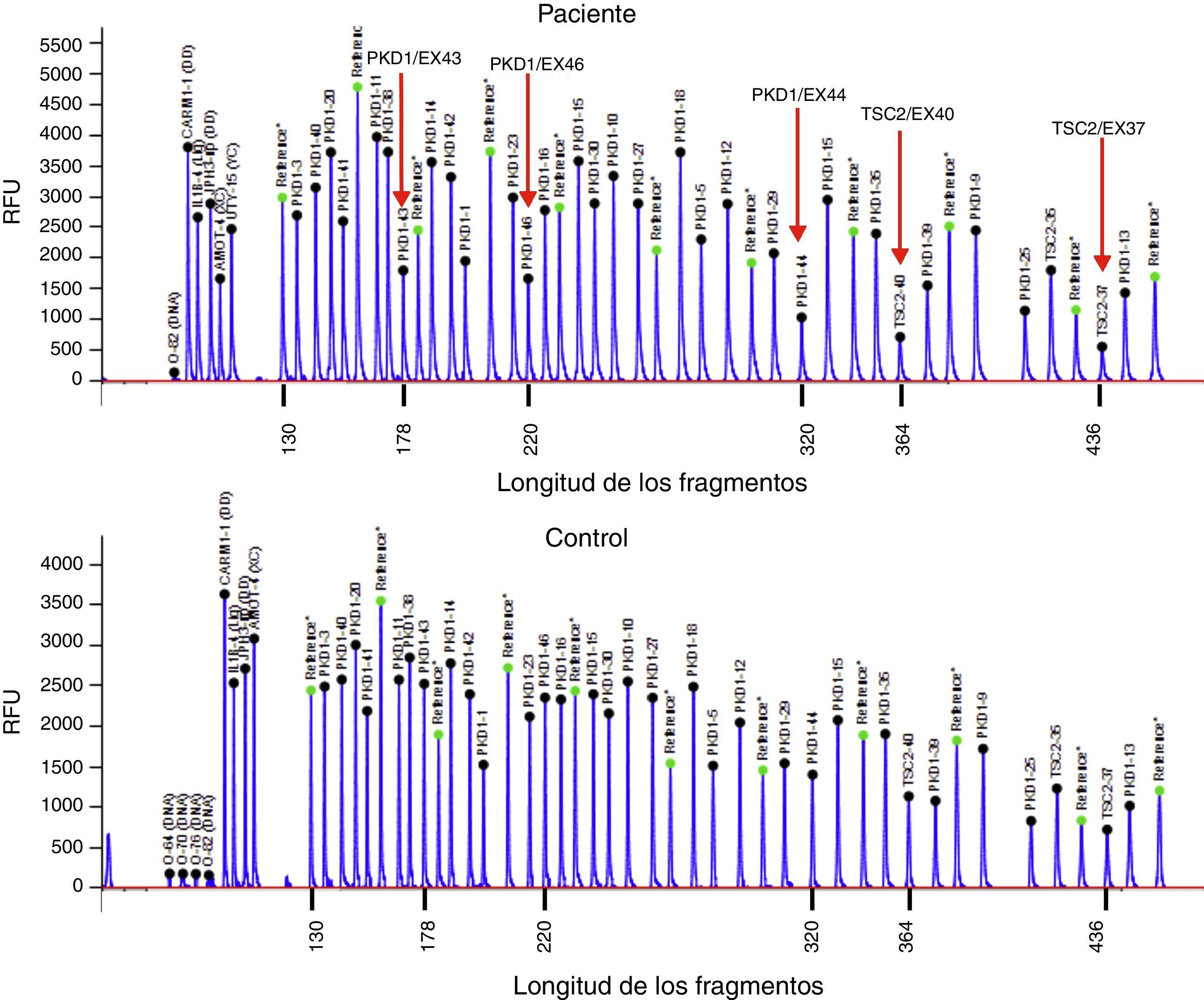
Deleción de genes contiguos TSC2/PKD1. Diagrama de resultados del análisis de deleciones/duplicaciones mediante la técnica de Multiplex Ligation-dependent Probe Amplification (MLPA) del paciente con SGC (panel superior) y de un individuo control (panel inferior). Las flechas rojas indican la deleción de los exones 43 al 46 del gen PKD1 (secuencias de referencia de GenBank: NM_001009944.2; NP_001009944.2): c.11713-?_12909+?del; p.(Ser1555fs)) y los exones 37 al 42 del gen TSC2 (NM_000548; NP_000539): c.4663-?_5421+?del; p.(Val3905fs)). Nomenclatura según la Human Genome Variation Society (http://www.hgvs.org/mutnomen/).
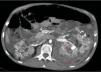
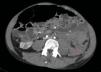
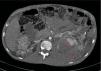
A los 32 años, acudió a urgencias por un cuadro digestivo de 3 días de evolución caracterizado por dolor abdominal, distensión y deposiciones líquidas sin productos patológicos. El cuadro clínico fue autolimitado, pero el paciente permanecía asténico. Al examen físico se encontraba afebril, con estabilidad hemodinámica, dolor difuso a la palpación abdominal sin signos de peritonismo. La analítica destacaba hemoglobina 71g/l (VN: 140-180g/l), hematocrito 0.22 L/L (VN: 0,40-0,52) sin criterios de hemólisis. Los parámetros de sepsis se encontraban en el rango de normalidad, la función renal permaneció estable con acidosis metabólica (creatinina 232 umol/l, filtrado glomerular [CKD-EPI] 30ml/min/1,73m2, bicarbonato 16 mEq/l). La ecografía abdominal mostraba líquido libre perihepático, por lo cual se completó el estudio con una TC abdominal. Se visualizó un hematoma retroperitoneal izquierdo (60×62×52mm) así como múltiples AML con microaneurismas bilaterales: el mayor era de 10mm a nivel del riñón izquierdo nativo (figs. 2–4). Se administraron 2 concentrados de hematíes, profilaxis antibiótica y se mantuvo conducta expectante. Dada la correcta evolución del paciente, fue dado de alta hospitalaria.
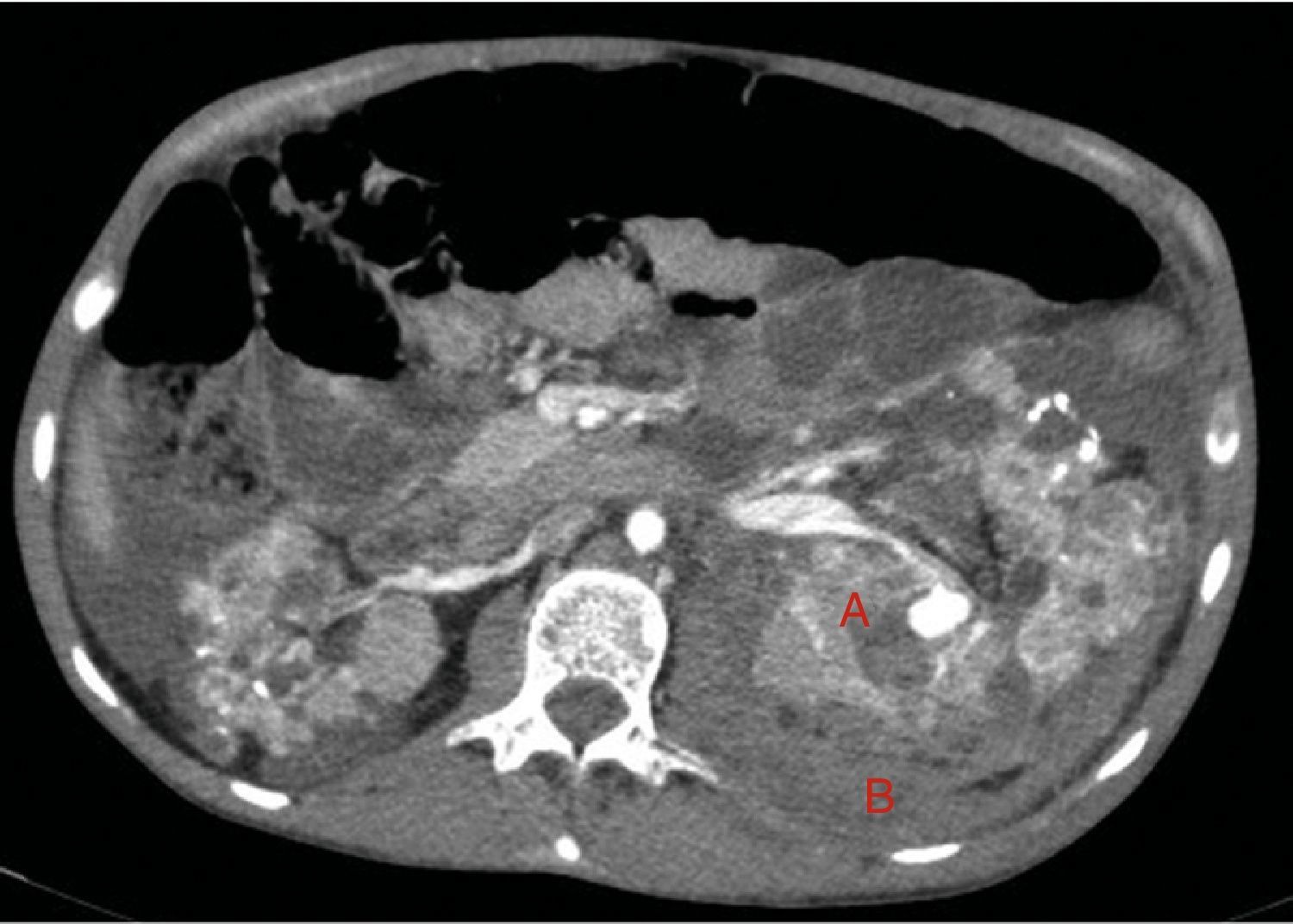
Riñones nativos con múltiples quistes, algunos con paredes calcificadas. Lesiones sugestivas de angiomiolipomas con múltiples aneurismas bilaterales, el mayor de ellos de 10mm en tercio superior del riñón izquierdo (A). Sangrado retroperitoneal izquierdo (B) que se extiende caudalmente por el espacio pararrenal posterior.
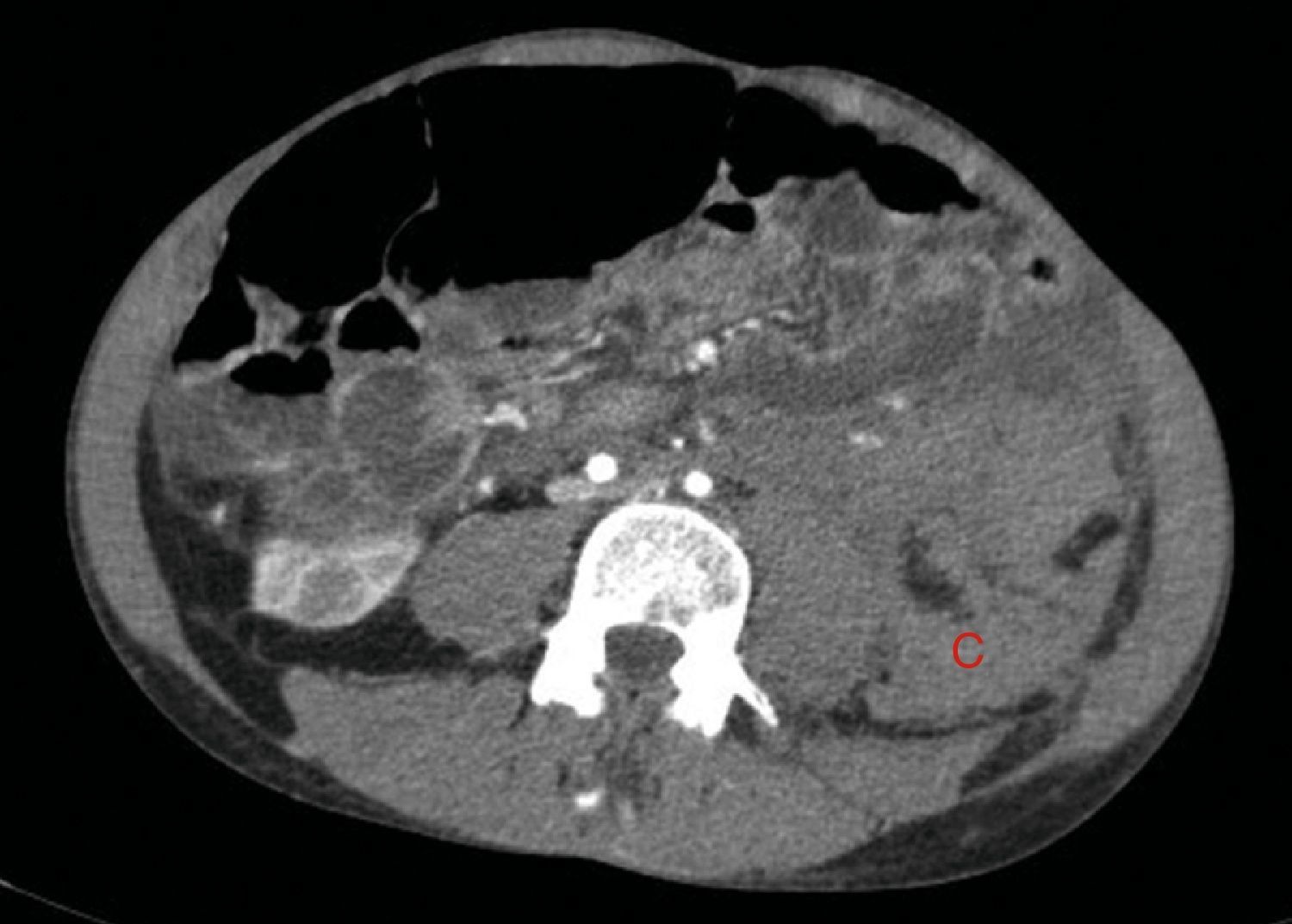
El sangrado descrito en la figura 2 conforma un voluminoso hematoma distal al polo renal inferior (C).
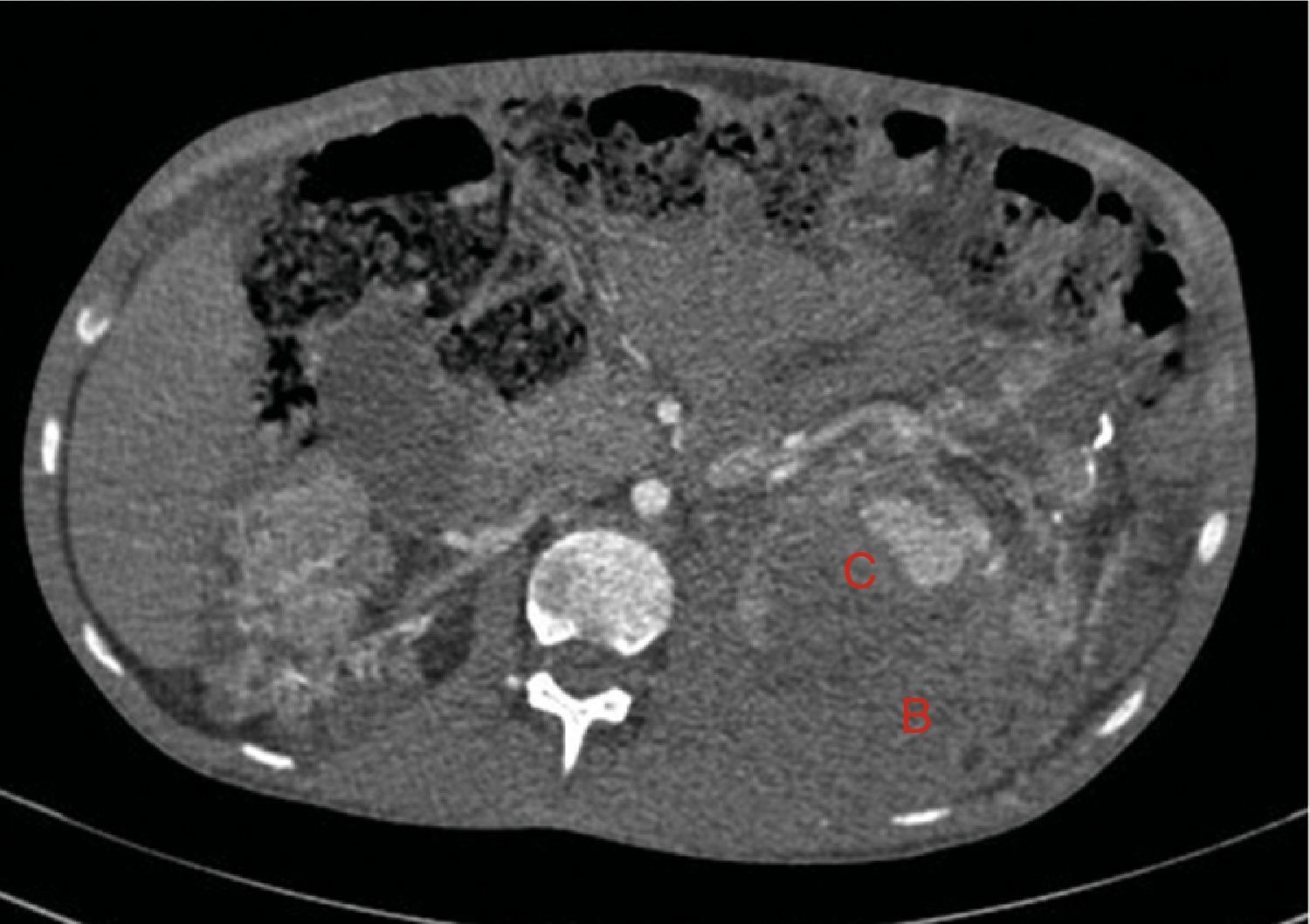
A los 15 días consultó por febrícula y dolor abdominal, sin clínica de bacteriemia ni foco infeccioso evidente. La analítica mostraba estabilidad en las cifras de hemoglobina, sin leucocitosis, pero destacaba PCR: 174 (VN: <10), aumento de creatinina 300umol/l y filtrado glomerular (CKD-EPI) 22ml/min/1,73 m2. Los hemocultivos y urinocultivo fueron negativos. Las radiografías de tórax y abdomen fueron normales. Se realizó TC abdominal a las 48 h por anemización (hemoglobina 69g/l, hematocrito 0.21 L/L), con aumento del hematoma retroperitoneal izquierdo (80×170×100mm) e incremento del tamaño del pseudoaneurisma del tercio superior del riñón izquierdo respecto al estudio previo (fig. 5). Como tratamiento se optó por la embolización selectiva de la arteria renal izquierda, sin incidencias. Se transfundieron 2 concentrados de hematíes. Como profilaxis para el síndrome postembolización se aumentaron los corticoides que tomaba a 0,5mg/kg y se pautó analgesia. Así mismo, se inició tratamiento antibiótico con una cefalosporina de tercera generación de forma empírica. La evolución fue correcta y el paciente fue dado de alta a los 5 días.
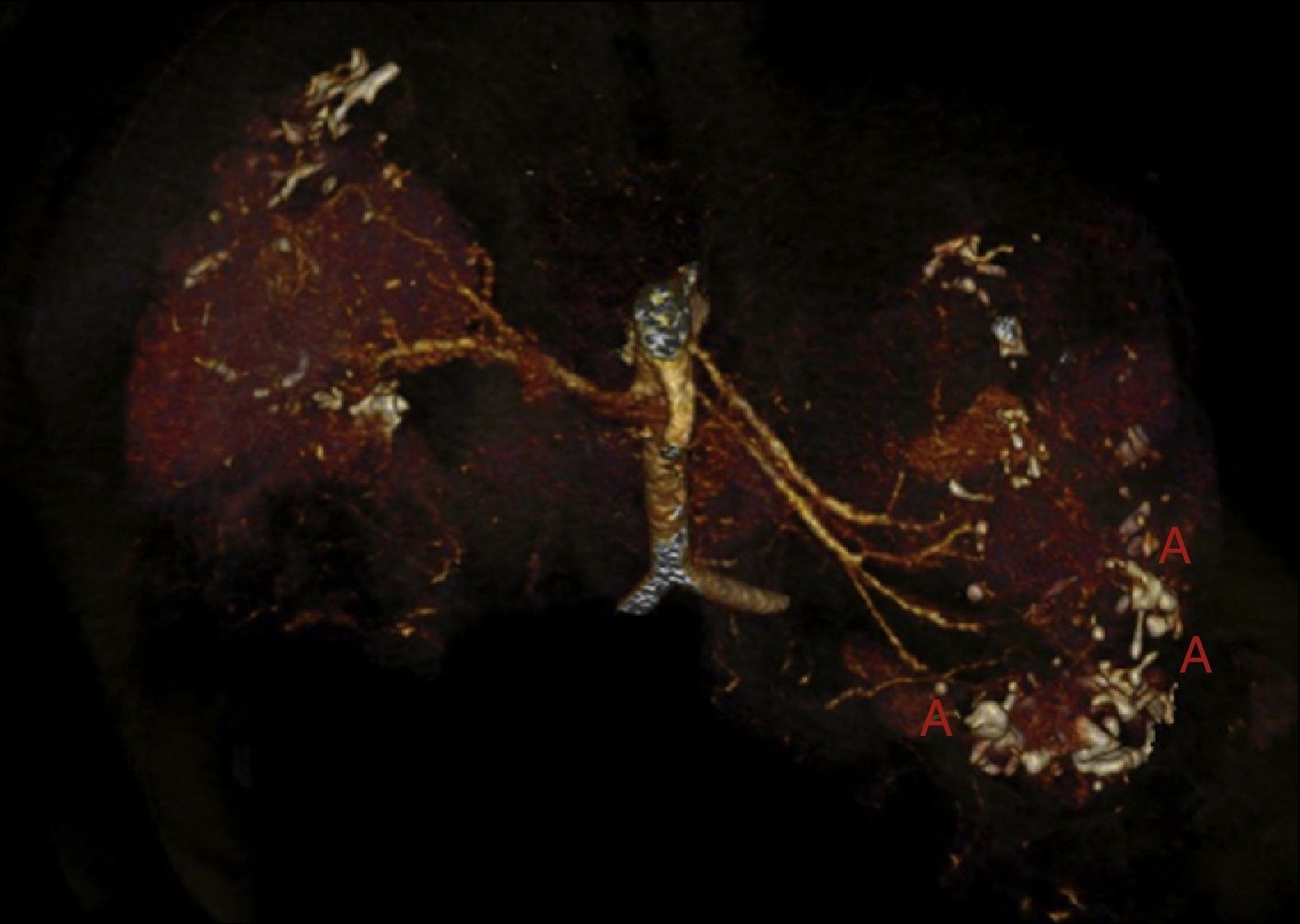
Es poco habitual que un individuo presente mutaciones en 2genes diferentes y, por lo tanto, presente ambas enfermedades. La excepción a esta rareza son los SGC como en el caso de la deleción de los genes TSC2/PKD17.
Alrededor de un 5% de los pacientes con ET presentan al mismo tiempo PQRAD, debido a una deleción que implica a los genes TSC2 y PKD111. La primera causa de muerte en la ET son las complicaciones neurológicas y la segunda son las complicaciones renales11. Los pacientes con ET pueden presentar quistes renales, pero la manifestación más frecuente y causante de mayor morbilidad son los AML. Estos tumores benignos pueden sangrar, con mayor riesgo cuanto más grandes sean. Es muy importante diagnosticar la presencia de microaneurismas en los AML porque supone un alto riesgo de rotura12.
La rapamicina y el everolimus son inmunosupresores que tienen la capacidad de bloquear la actividad de mTOR produciendo un efecto antiproliferativo directo, inhibiendo la actividad de células T y disminuyendo el riesgo de angiogénesis por disminución de los niveles de factor de crecimiento endotelial vascular. De esta manera, se reduce el tamaño de los AML, fibromas, astrocitomas y, probablemente, la frecuencia de crisis epilépticas. Cabrera et al. reportaron a 17 pacientes con ET tratados con rapamicina: el 100% de los casos presentaron disminución del tamaño de los AML superior al 50% al año de tratamiento. Dicha disminución de tamaño permaneció estable durante el 1.er y 2.° año de tratamiento19,20. El estudio aleatorizado y multicéntrico EXIST-2, con una muestra mayor de 118 participantes, demostró una gran eficacia de everolimus en la disminución del volumen de los AML, con ausencia de sangrados15,21. Otros estudios apoyan también el beneficio de los inhibidores de mTOR en el tratamiento de los AML22,23. Por estos motivos, serían fármacos de elección en pacientes trasplantados con ET.
Con estos datos, creemos que nuestro paciente podría haberse beneficiado en el momento del trasplante renal del tratamiento con inhibidores de mTOR. Este fármaco actúa como inmunosupresor y reductor tumoral en la ET disminuyendo el riesgo de rotura y hemorragia severa. La tolerancia de los inhibidores de mTOR no siempre es buena, y deben ser retirados hasta en un porcentaje significativo de los pacientes por efectos indeseables y mala tolerancia. También se sabe que su introducción con filtrados glomerulares disminuidos y, especialmente, cuando existe proteinuria superior a medio gramo se asocia a deterioro funcional del injerto renal. En este caso no se administró porque no se conocía la relación de los inhibidores de mTOR con la ET cuando el paciente se trasplantó. En el momento en que se conoció dicho efecto en la ET, dado el deterioro de función renal con filtrado glomerular por debajo de 30ml/min/173 m2 y cierto grado de proteinuria debido a disfunción crónica del injerto, no se estimó adecuado administrarlo. Por otra parte, no se contempló la posibilidad de que, tras 17 años de trasplante, los riñones propios tuviesen perfusión renal suficiente como para ocasionar un sangrado. Este caso confirma que, a pesar de tratarse de pacientes trasplantados o en diálisis, el riesgo de sangrado asociado a los AML se mantiene, por lo que la utilización de inhibidores de mTOR en un paciente trasplantado renal con AML por ET podría mantener la funcionalidad del injerto renal evitando el rechazo y disminuyendo el tamaño de los AML, con menor riesgo de complicaciones. Debemos destacar que el riñon derecho de nuestro paciente también presentaba múltiples AML, el mayor de los cuales era de 5cm. Pero como se trataba de un paciente lábil, institucionalizado y con delicada calidad de vida, se intentó ser lo menos agresivos posible en ese momento para limitarnos al tratamiento del sangrado agudo. De todos modos, queda pendiente realizar la embolización del riñón derecho. En este caso, se optó por la embolización renal, por ser un procedimiento menos invasivo y con menos riesgo de complicaciones que una intervención quirúrgica como la nefrectomía.
En base en nuestra experiencia, se recomienda el seguimiento mediante angio-TC o resonancia magnética nuclear de estos pacientes, con el fin de detectar AML con riesgo de sangrado y valorar la embolización o nefrectomía profiláctica cuando no sea adecuada la utilización de inhibidores de mTOR. El presente caso de ET confirma que la embolización es una técnica perfectamente aceptable y segura en AML renales sangrantes o con riesgo de sangrado que no puedan recibir inhibidores de mTOR.
En conclusión, aun en pacientes con ET que reciban terapia renal sustitutiva, se debe seguir realizando controles periódicos de sus riñones propios. En caso de observarse AML de tamaño superior a 3cm se aconseja, si es factible, usar inhibidores de mTOR en pacientes trasplantados o con embolización renal o nefrectomía tanto en pacientes en diálisis como trasplantados.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



