


Membranoproliferative glomerulonephritis (MPGN type I, II and III) was reclassified in 2013 as MPGN and C3 glomerulopathy (C3G) based on the complement system activation mechanism.
ObjectivesTo evaluate whether C4d, a component of the classical pathway, could be a diagnostic tool in differentiating between MPGN and C3G.
MethodsWe conducted a retrospective study of 15 MPGN type I, II and III and 13 minimal change disease (MCD) patients diagnosed between 2000 and 2012. C4d staining using the peroxidase method was employed.
ResultsUsing the 2013 C3G consensus classification, the 15 MPGN types I, II and III biopsies were re-classified as MPGN (8) and C3G (7). Following C4d staining, of the 8 biopsies diagnosed as MPGN, 4 had classical pathway involvement [C1q (+), C3 (+), C4d (+)]; two had lectin pathway involvement [C1q (−), C3 (+), C4d (+)]; and, two were reclassified as C3G because the absence of C4d and C1q suggested the presence of the alternative pathway [C1q (−), C3 (+), C4d (−)]. Three of the seven C3G biopsies presented classical pathway involvement and were reclassified as MPGN. The alternative pathway was present in one of the other 4 biopsies considered to be C3G. Two C3G biopsies involved the lectin pathway and the one case of dense deposit disease had lectin pathway involvement.
ConclusionsC4d staining may help to differentiate between MPGN and C3G. In addition, the lectin pathway could play a role in the pathogenesis of these glomerulopathies.
La glomerulonefritis membranoproliferativa (GnMP, tipo I, II y III) fue reclasificada en 2013 como GnMP y glomerulopatía C3 (GC3) en base al mecanismo que activa el sistema del complemento.
ObjetivosEvaluar si C4d, componente de la vía clásica, puede diferenciar GnMP y GC3.
MétodosEstudio retrospectivo incluyendo 15 pacientes con GnMP (tipo I, II y III) y 13 con enfermedad de cambios mínimos (CM) diagnosticados entre 2000 y 2012. Realizamos tinción renal con C4d mediante el método de la peroxidasa.
ResultadosEn base a la definición de GC3 consensuada en 2013, las 15 biopsias diagnosticadas como GnMP se reclasificaron como GnMP y GC3 en 8 y 7 casos respectivamente. Tras la tinción de C4d; de las 8 biopsias diagnosticadas como GnMP, 4 mostraron activación de la vía clásica [C1q (+), C3 (+), C4d (+)], 2 activación de la vía de las lectinas (VL) [C1q (-), C3 (+), C4d (+)]; y 2 fueron reclasificadas como GC3 dada la ausencia de C4d y C1q sugiriendo participación de la vía alternativa [C1q (-), C3 (+), C4d (-)]. Tres de 7 biopsias diagnosticadas de GC3fueron reclasificadas como GnMP debido a las presencia de activación de la vía clásica. La vía alternativa estuvo presente in 1 de las otras 4 biopsias consideradas GC3. VL estuvo activada en 2 de biopsias diagnosticadas de GC3 y en el único caso de enfermedad de depósitos densos.
ConclusionesC4d puede ayudar a diferenciar GnMP y GC3. La VL podría jugar un papel en la patogenia de estas glomerulopatías.
Membranoproliferative glomerulonephritis is a pattern of glomerular injury that results from several distinct etiologies of glomerular disease. Since the 1970s, the initial classification for MPGN has included MPGN type I, II (dense deposit disease or DDD), and III based on light microscopy (LM), immunofluorescence (IF), and electron microscopy (EM).1–5
Membranoproliferative glomerulonephritis is characterized on LM by mesangial and endothelial hypercellularity, increased mesangial matrix, and thickening of the glomerular capillary wall, often giving it an accentuated lobular appearance. By IF, there is granular capillary wall deposition of (IgG and/or IgM) with complement C3 in MPGN type I. Immunoglobulins are usually absent in the glomeruli of DDD and MPGN type III. On EM, electron dense deposits are usually located in the subendothelial region in MPGN type I, intramembranous location in DDD, and subendothelial, intramembranous and subepithelial areas in MPGN type III. DDD is defined by its characteristic osmiophilic intramembranous deposits on EM and predominance of complement C3 on IF.
The first C3 Glomerulopathy6 consensus introduced a classification for membranoproliferative glomerulonephritis based on the pathogenesis involving the complement cascade. MPGN associated with deposition of immunoglobulins and complement (previously called MPGN type I or III) is believed to be immune complex mediated and; therefore, activates the classical complement pathway. Cases with predominant C3 deposition with little or no immunoglobulin staining implies complement activation via the alternative pathway and are designated C3 glomerulopathy (C3G).7–18 Using a strict approach, C3G has been defined as “dominant C3 of ≥2 orders of magnitude of intensity by IF greater than any other immune reactant”.14 C3G is subdivided into DDD and C3 glomerulonephritis (C3GN). DDD continues to be characterized by dark, osmophilic intramembranous deposits on EM and predominance of complement C3.19 C3GN now incorporates some of those that were previously designated as MPGN types I and III.
As previously mentioned, the pathogenesis of MPGN and C3G involves the activation of the classical and alternative pathways, respectively. The complement system consists of three pathways: classical, lectin, and alternative. All three pathways end up activating C3, which then progresses to form the membrane attack complex (C5-C9) to induce localized cellular injury and inflammation. 20,21 The classical pathway is initiated by immunoglobulins and involves the early complement components of C1q, C2 and C4. The lectin pathway is initiated by mannose-binding lectin or ficolins which activate C2 and C4. No C1q is involved in the lectin pathway. The classical and lectin pathways end result is the formation of C3 convertase (C4b2b). The alternative pathway has a spontaneous activation of C3 by hydrolysis of its thioester bond that initiates the complement cascade and does not involve the early components of the cascade (C1q, C2 and C4). As the result of the cleavage of C4 in the lectin and classical pathways, C4d is formed. Since C4d has an internal thioester bond, it has the ability to form a covalent bond to cell surfaces. With C4d being tightly anchored to the tissue site, it is an ideal marker of inflammation or injury.22
Since no significant C4b or its metabolite C4d is formed in the alternative pathway, we have used C4d immunohistochemistry as a method to screen for pathways of complement activation involved in the pathogenesis. Positive glomerular capillary wall immunoreactivity for C4d would favor involvement of the classical complement pathway and negative C4d staining would favor involvement of the alternative pathway
Materials and methodsPatient selectionThis retrospective study included both adult and pediatric patients who had the 1970 original diagnosis of MPGN type I, II, III. Minimal change disease (MCD) served as a comparison control group. Patients were identified from the Informatics for Integrating Biology and the Bedside Query & Analysis Tool software based on the ICD-9 codes. ICD-9 codes 581.2, 582.2, and 583.2 were used to identify patients with MPGN. ICD-9 code 581.3 was used to identify patients with MCD. The patients were seen at the University of Florida (UF) from January 2000 to December 2012. Patients with de novo and recurrent MPGN type I, II, III in renal transplant were also included, along with a heterogeneous mixture of primary and secondary causes of MPGN (atypical hemolytic uremic syndrome, cryoglobulinemia, hepatitis C, and monoclonal gammopathy of unknown significance). Pertinent clinical data were recorded. The study was approved by the Institutional Review Board office at UF.
C4d ImmunohistochemistryFormalin-fixed paraffin-embedded tissue sections were stained using the rabbit anti-human C4d polyclonal antibody (American Research Products, Inc, Waltham, MA, catalog number 12-5000) via an immunoperoxidase method established at our institution (Procedure: XT ultraView DAB v3).
Four micron thick paraffin slides were placed on plus slides, dried for 2h in a 60°C oven and placed on the Ventana Benchmark automated immunostainer where they were dewaxed and heat induced epitope retrieval was performed with Ventana's CC1 retrieval solution for 30min at 95–100°C. Primary antibody, anti-human C4D (1:20 dilution) was applied to sections at 37°C for 32min. Immunoreactivity was visualized by using the Ultra View DAB detection kit (Ventana Medical Systems, Inc. Tucson, AZ). Slides were counterstained with hematoxylin, dehydrated, cleared, and mounted with permanent mounting media.
Glomerular staining results for C1q and C3, performed by standard immunofluorescence methodology, were collected during the review of the pathology reports.
Pathology reviewThe histological sections were reviewed by all authors who were blinded to any identifiers during the reviews. To include patients in this study, any number of glomeruli was accepted. First, the cases were categorized based on their original pathology report and subsequently classified based on the new 2013 C3G consensus report in which glomerular C3 staining of at least >2 orders of magnitude of intensity by IF greater than any other immune reactant was considered dominant and indicative of C3G. DDD was characterized by its classic osmophilic intramembranous deposits on EM. Finally, the cases were sorted based on the C4d immunoreactivity results.
Scoring of slidesOnly peripheral capillary loop staining for C4d was scored. To score glomeruli as positive or negative staining, we relied on both the intensity and distribution of the immunoreactivity. Intensity of C4d staining in the capillary loop was based on a scale of 0 to 3+ staining. The distribution was either global or segmental capillary loop staining. Glomeruli with global 0 to trace C4d staining were considered negative. Glomeruli regardless of intensity with less than 15% segmental C4d staining were also considered negative. Sclerotic, scarred, or collapsed glomerulus, whether global or segmental which may have IgM, C3 and/or C1q trapping, were not scored. Global capillary (GC) loop staining had ≥50% of the glomerulus staining with greater than 1+ intensity. Segmental capillary (SC) loop staining had <50% of the glomerulus staining with greater than 1+ intensity. Diffuse C4d staining was defined as ≥50% of the total glomeruli staining, and focal C4d staining was defined as <50% of the total glomeruli staining. Any disagreement on biopsy classification or intensity staining was resolved by reviewing the pathology slides as a group and agreeing with the majority.
StatisticsStatistics were performed using Mann–Whitney and chi-squared tests.
ResultsAfter the initial screening, 23 MCD and 32 MPGN type I, II, III patients were selected. Of the MCD patients, those with no reported IF (n=4) were excluded. Of the MPGN patients, those with no reported IF (n=7), EM (n=2), or neither IF and EM (n=2) were excluded. After refacing the paraffin block of the original biopsy samples, 6 MPGN and 6 MCD patients had no glomeruli for C4d immunohistochemical (IHC) staining. Therefore, our study included a final sample of 13 MCD, 14 MPGN (type I and III) and 1 DDD patient.
Patients and laboratory findings (Table 1)The MCD patients were 3–25 years old (average=11.5 years), with 3 males and 10 females. The MPGN type I, II, III patients were 11–66 years old (average=36.5 years), with 5 males and 10 females.
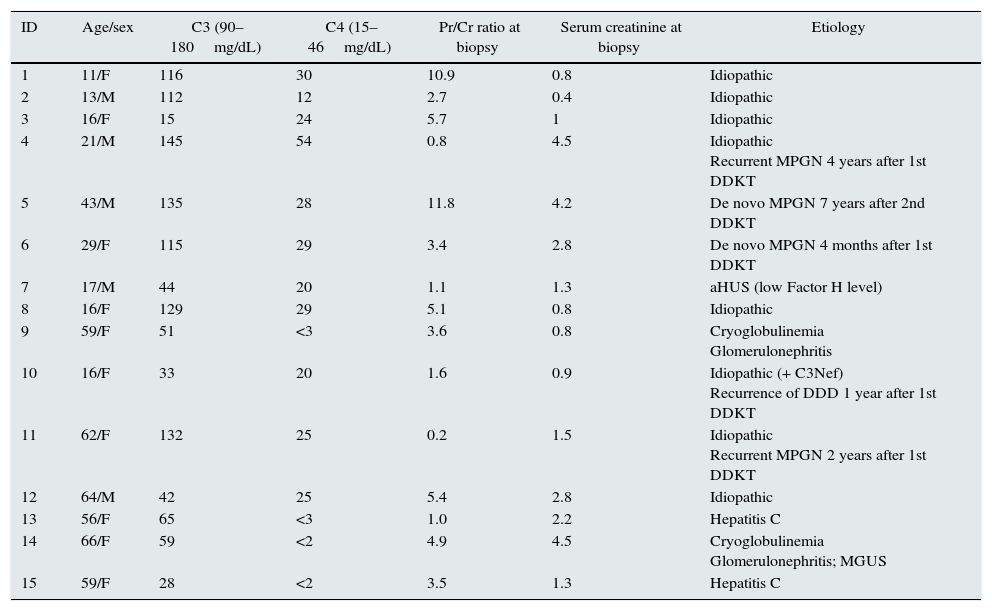
Pathological and clinical data of patients.
| ID | Age/sex | C3 (90–180mg/dL) | C4 (15–46mg/dL) | Pr/Cr ratio at biopsy | Serum creatinine at biopsy | Etiology |
|---|---|---|---|---|---|---|
| 1 | 11/F | 116 | 30 | 10.9 | 0.8 | Idiopathic |
| 2 | 13/M | 112 | 12 | 2.7 | 0.4 | Idiopathic |
| 3 | 16/F | 15 | 24 | 5.7 | 1 | Idiopathic |
| 4 | 21/M | 145 | 54 | 0.8 | 4.5 | Idiopathic Recurrent MPGN 4 years after 1st DDKT |
| 5 | 43/M | 135 | 28 | 11.8 | 4.2 | De novo MPGN 7 years after 2nd DDKT |
| 6 | 29/F | 115 | 29 | 3.4 | 2.8 | De novo MPGN 4 months after 1st DDKT |
| 7 | 17/M | 44 | 20 | 1.1 | 1.3 | aHUS (low Factor H level) |
| 8 | 16/F | 129 | 29 | 5.1 | 0.8 | Idiopathic |
| 9 | 59/F | 51 | <3 | 3.6 | 0.8 | Cryoglobulinemia Glomerulonephritis |
| 10 | 16/F | 33 | 20 | 1.6 | 0.9 | Idiopathic (+ C3Nef) Recurrence of DDD 1 year after 1st DDKT |
| 11 | 62/F | 132 | 25 | 0.2 | 1.5 | Idiopathic Recurrent MPGN 2 years after 1st DDKT |
| 12 | 64/M | 42 | 25 | 5.4 | 2.8 | Idiopathic |
| 13 | 56/F | 65 | <3 | 1.0 | 2.2 | Hepatitis C |
| 14 | 66/F | 59 | <2 | 4.9 | 4.5 | Cryoglobulinemia Glomerulonephritis; MGUS |
| 15 | 59/F | 28 | <2 | 3.5 | 1.3 | Hepatitis C |
Note: MPGN, membranoproliferative glomerulonephritis; DDKT, deceased donor kidney transplant; LRKT, living related kidney transplant; aHUS, atypical hemolytic uremic syndrome; C3Nef, C3 nephritic factor; MGUS, monoclonal gammopathy of unknown significance.
The etiologies of MPGN were: idiopathic MPGN (n=8), de novo MGPN in transplant (n=2), atypical hemolytic uremic syndrome (n=1), Hepatitis C (n=2), cryoglobulinemia (n=1), and cryoglobulinemia with MGUS (monoclonal gammopathy of unknown significance) (n=1). The DDD patient tested positive for C3 nephritic factor and had recurrence of DDD two years after the transplant. Only 1 of 5 (ID 4) renal transplant biopsies reported mild allograft rejection with C4d staining.
C4d ImmunohistochemistryMCD: glomerular staining with C4d immunohistochemistryTwelve biopsies had negative C4d glomerular capillary loop staining. Only one biopsy had diffuse glomerular staining. Of the 17 glomeruli available in this biopsy, 8 had segmental capillary loop (SC) staining of less than 25% per glomerulus, 1 glomeruli had global capillary loop (GC) staining, and 8 glomeruli had negative staining. Overall, there were a total of 127 glomeruli in the MCD biopsy cases, of which only 9 glomeruli stained with C4d (7%).
MPGN and C3G: glomerular staining with C4d immunohistochemistryTwelve biopsies (80%) had C4d glomerular staining (see Table 2). Eleven of 12 biopsies (92%) had diffuse glomerular GC staining including the DDD case. Two biopsies (ID 3 and 12) had diffuse GC staining with some focal glomerular C4d negativity (1 of 13 glomeruli and 3 of 13 glomeruli, respectively). One case (ID 7) had focal segmental distribution with 2 of 7 glomeruli showing SC staining and 5 of 7 glomeruli negative. Three cases (ID 13, 14, 15) had completely negative glomerular capillary loop staining.
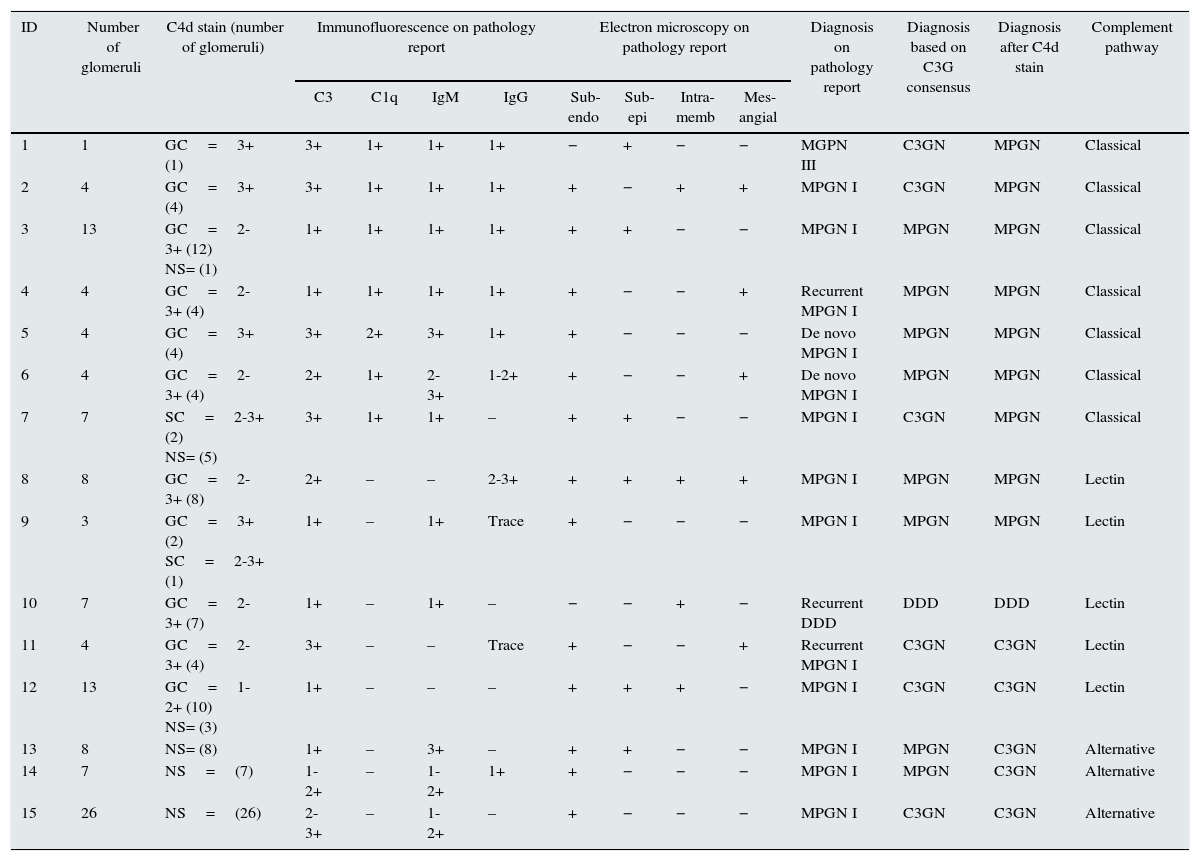
C4d Immunohistochemistry stain in MGPN and C3G.
| ID | Number of glomeruli | C4d stain (number of glomeruli) | Immunofluorescence on pathology report | Electron microscopy on pathology report | Diagnosis on pathology report | Diagnosis based on C3G consensus | Diagnosis after C4d stain | Complement pathway | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C3 | C1q | IgM | IgG | Sub-endo | Sub-epi | Intra-memb | Mes-angial | |||||||
| 1 | 1 | GC=3+ (1) | 3+ | 1+ | 1+ | 1+ | − | + | − | − | MGPN III | C3GN | MPGN | Classical |
| 2 | 4 | GC=3+ (4) | 3+ | 1+ | 1+ | 1+ | + | − | + | + | MPGN I | C3GN | MPGN | Classical |
| 3 | 13 | GC=2-3+ (12) NS= (1) | 1+ | 1+ | 1+ | 1+ | + | + | − | − | MPGN I | MPGN | MPGN | Classical |
| 4 | 4 | GC=2-3+ (4) | 1+ | 1+ | 1+ | 1+ | + | − | − | + | Recurrent MPGN I | MPGN | MPGN | Classical |
| 5 | 4 | GC=3+ (4) | 3+ | 2+ | 3+ | 1+ | + | − | − | − | De novo MPGN I | MPGN | MPGN | Classical |
| 6 | 4 | GC=2-3+ (4) | 2+ | 1+ | 2-3+ | 1-2+ | + | − | − | + | De novo MPGN I | MPGN | MPGN | Classical |
| 7 | 7 | SC=2-3+ (2) NS= (5) | 3+ | 1+ | 1+ | – | + | + | − | − | MPGN I | C3GN | MPGN | Classical |
| 8 | 8 | GC=2-3+ (8) | 2+ | – | – | 2-3+ | + | + | + | + | MPGN I | MPGN | MPGN | Lectin |
| 9 | 3 | GC=3+ (2) SC=2-3+ (1) | 1+ | – | 1+ | Trace | + | − | − | − | MPGN I | MPGN | MPGN | Lectin |
| 10 | 7 | GC=2-3+ (7) | 1+ | – | 1+ | – | − | − | + | − | Recurrent DDD | DDD | DDD | Lectin |
| 11 | 4 | GC=2-3+ (4) | 3+ | – | – | Trace | + | − | − | + | Recurrent MPGN I | C3GN | C3GN | Lectin |
| 12 | 13 | GC=1-2+ (10) NS= (3) | 1+ | – | – | – | + | + | + | − | MPGN I | C3GN | C3GN | Lectin |
| 13 | 8 | NS= (8) | 1+ | – | 3+ | – | + | + | − | − | MPGN I | MPGN | C3GN | Alternative |
| 14 | 7 | NS=(7) | 1-2+ | – | 1-2+ | 1+ | + | − | − | − | MPGN I | MPGN | C3GN | Alternative |
| 15 | 26 | NS=(26) | 2-3+ | – | 1-2+ | – | + | − | − | − | MPGN I | C3GN | C3GN | Alternative |
Note: Subendo, subendothelial deposits; Subepi, subepithelial deposits; Intramemb, intramembranous deposits.
GC,global capillary loop (>50% glomerular stain); SC,segmental capillary loop (<50% glomerular stain); NS,no glomerular stain;; MPGN, membranoproliferative glomerulonephritis); C3GN, C3 glomerulonephritis; DDD, dense deposit disease.
Overall, 63 of 113 glomeruli in the MPGN and C3G groups stained with C4d (56%). Between the MPGN and C3G groups, the MPGN had more glomerular staining (88% vs. 32%). The difference in C4d staining between MCD, MPGN and C3G was also statistically significant (p<0.001). The intensity of the staining was variable from 1+ to 3+, with a predominance of 3+ staining when compared to the MCD biopsies.
MPGN: classification using the C3G consensus criteriaThe 15 MPGN type I, II, III cases based on the original pathology report were reclassified according to the C3G consensus criteria after reviewing the C3 IF results (see Table 2). Six patients were designated as C3GN and eight patients were designated as MPGN. One patient despite the lack of dominant C3 IF pattern was classified as DDD because she had a characteristic electron-dense intramembranous appearance on EM.
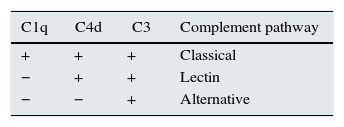
MPGN: classification using C4d and C1q for complement pathway identificationWe define the complement pathway activation using the combined results of the C4d and C1q stains (see Tables 3 and 4; Fig. 1). If C1q and C4d were positive, the classical pathway was suggested; if both Clq and C4d were negative, the alternative pathway was suggested; if C1q was negative and C4d was positive, then the lectin pathway was suggested.
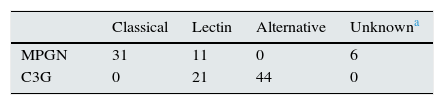
Number of glomeruli involved in each specific complement pathway.
| Classical | Lectin | Alternative | Unknowna | |
|---|---|---|---|---|
| MPGN | 31 | 11 | 0 | 6 |
| C3G | 0 | 21 | 44 | 0 |
 that is consistent with a membranoproliferative pattern along with immunofluorescence (IF) and electron microscopy (EM) pattern defines MPGN and C3G. By using C4d immunohistochemistry, C1q and C3 on IF, the involvement of a particular complement pathway can be suggested. Minimal change disease (MCD) biopsy serves as a control. There is no detection of glomerular capillary loop C4d stain (a), no presence of glomerular C1q (d) or C3 (g) by IF, and on EM (j) there is podocyte effacement with no electron dense deposits. MPGN (classical) phenotype shows predominant global capillary loop C4d stain (b), positive glomerular C1q (e) and positive C3 (h) on IF. The EM (k) shows presence of subendothelial electron dense deposits. MPGN (lectin) phenotype shows predominance of global capillary loop and mesangial C4d staining (c), negative glomerular C1q (f) and positive glomerular C3 (i) on IF. The EM (l) here shows presence of large subendothelial and mesangial deposits. DDD (lectin) phenotype shows dramatic linear global capillary loop C4d stain (m), negative glomerular C1q (p), and positive glomerular C3 (s) on IF. The EM (v) here shows the characteristic dark, linear osmophilic intramembranous electron dense deposits. C3GN (lectin) phenotype shows predominance of global capillary loop C4d stain (n), negative glomerular C1q (q), and positive glomerular C3 (t) on IF. The EM (w) here shows subendothelial deposits. C3GN (alternative) phenotype shows the no detection of capillary loop C4d stain, negative C1q, and positive C3. The EM shows (x) subendothelial and mesangial deposits.")
Biopsy classification involving the complement cascade based on C4d stain, IF, and EM. The light microscopy (not shown) that is consistent with a membranoproliferative pattern along with immunofluorescence (IF) and electron microscopy (EM) pattern defines MPGN and C3G. By using C4d immunohistochemistry, C1q and C3 on IF, the involvement of a particular complement pathway can be suggested.
Minimal change disease (MCD) biopsy serves as a control. There is no detection of glomerular capillary loop C4d stain (a), no presence of glomerular C1q (d) or C3 (g) by IF, and on EM (j) there is podocyte effacement with no electron dense deposits.
MPGN (classical) phenotype shows predominant global capillary loop C4d stain (b), positive glomerular C1q (e) and positive C3 (h) on IF. The EM (k) shows presence of subendothelial electron dense deposits.
MPGN (lectin) phenotype shows predominance of global capillary loop and mesangial C4d staining (c), negative glomerular C1q (f) and positive glomerular C3 (i) on IF. The EM (l) here shows presence of large subendothelial and mesangial deposits.
DDD (lectin) phenotype shows dramatic linear global capillary loop C4d stain (m), negative glomerular C1q (p), and positive glomerular C3 (s) on IF. The EM (v) here shows the characteristic dark, linear osmophilic intramembranous electron dense deposits.
C3GN (lectin) phenotype shows predominance of global capillary loop C4d stain (n), negative glomerular C1q (q), and positive glomerular C3 (t) on IF. The EM (w) here shows subendothelial deposits.
C3GN (alternative) phenotype shows the no detection of capillary loop C4d stain, negative C1q, and positive C3. The EM shows (x) subendothelial and mesangial deposits.
Of the 8 biopsies diagnosed with MPGN using the consensus criteria, 4 had classical pathway involvement due to glomerular staining with C1q (+), C3 (+), C4d (+); and, two biopsies had lectin pathway involvement due to C1q (−), C3 (+), C4d (+) pattern. Two biopsies in this group were reclassified as C3G because the absence of C4d and C1q suggested the presence of the alternative pathway complement.
Three of the six C3G biopsies had C4d and C1q positive staining, suggesting classical pathway and were reclassified as MPGN. The alternative pathway (C1q (−), C3 (+), C4d (−)) was present in one of the other 3 C3G biopsies. In the other two, it seems that the lectin pathway was involved (C1q (−), C3 (+), C4d (+)).
DiscussionSince the 1970s and until recently, MPGN types I, II and III have been defined according to their characteristic pathological features observed on LM, IF and EM. However, many of the heterogeneous lesions of MPGN “could not be satisfactorily placed within existing pathological descriptions based on morphology.6” To resolve this confusion, the first C3 Glomerulopathy in 2012 attempted to describe MPGN based on its pathogenesis, where MPGN involves the classical complement pathway and C3G involves the alternative complement pathway activation. Based on the above proposed pathogenesis of MPGN and C3G, we introduced glomerular C4d and C1q immunohistochemistry (IHC) stains as an approach to differentiate between MPGN entities. Neither the C4d stain nor the C1q stains, which are involved in the early part of the complement cascade, were included in the recent C3G consensus.
The C4d technique can be performed by either paraffin-IHC or frozen-IF methods.23 Both techniques have reports of intra- and inter-observer variability. We use the paraffin-IHC method for two main reasons. One it does not require a frozen sample. Second, the IF method, although more sensitive and rapid with a lower background noise, is not a permanent staining and does not work on paraffin sections.24 The largely negative C4d IHC staining in the glomerular capillary wall of MCD cases and the stark C4d staining difference between C3GN and MPGN indicated that our C4d technique was appropriate and accurate.
In our study, we observed that C4d IHC showed intense, diffuse >2+ glomerular capillary wall staining in majority of the MPGN glomeruli when compared to the predominant negative staining of C4d in the C3G glomeruli (p<0.0001). Recently, a new entity of C4 DDD was reported where extensive work-up revealed renal deposits of C4d with “absence of alternative pathway activation”.25 Similar finding in our DDD patient supports that a pathway besides the presumed alternative pathway, such as the lectin pathway or C-reactive protein, may be involved in the pathogenesis. Although C-reactive protein can activate C4, it generally does not lead to full complement pathway activation.23
A recent study by Sethi et al.26 reported a high sensitivity and specificity for negative C4d (93% and 100%, respectively) and negative C1q (97% and 63%, respectively) in C3G versus immune complex-mediated GN (MPGN, SLE, IgA, membranous, fibrillary GN). Thus, Sethi's and our studies suggest that staining for C4d is valuable in differentiating C3G from MPGN.
Based on our complement composite profile of C1q, C3 and C4 staining, the presence of both C1q and C4d made a stronger case for the involvement of the classical pathway in seven biopsies, all of which were classified as MPGN. The absence of C1q and presence of C4d was suggestive of the lectin pathway in five biopsies, two were classified as MPGN and three as C3G. The absence of both C1q and C4d was more suggestive of the alternative pathway in three biopsies, all of which were C3G. Similar to Sethi et al.26 our results also emphasize that the lectin pathway may be common to both MPGN and C3G.
These results indicate that the pathogenesis in MPGN and C3G is more complex than previously thought, and that the pathogenesis proposed by C3G consensus may warrant further investigation. In fact, this study highlights the importance of a more prospective, comprehensive and larger study that includes further complement profile testing and confirm the presence of lectin pathway to formally elucidate the pathogenesis of MPGN and C3G.
It is also important to note that, in this study, the serum C4 level was variable among the MPGN and C3G groups. C4 was low in two of nine MPGN patients and in three of six C3G patients. Of the three C3GN patients, two had Hepatitis C and one had MGUS/cryoglobulinemia glomerulonephritis. It is believed that C4 is low in such patients due to persistent antigenemia and/or antigen-antibody immune complex deposition in the glomerulus.27-31 However, in the absence of glomerular C4d staining and absent cryoglobulins on renal biopsy, the low serum C4 may be related to extra-renal systemic consumption of the complements and not due to direct glomerular deposits. Another possibility for the low complement is hyposynthesis in the liver.32 Hence, the low serum C4 level may falsely reassure one of the classical pathway activation. It is also suggested that a primary trigger can lead to unregulated alternative pathway activation that overwhelms the compensatory regulatory mechanism, triggering glomerular deposition of complement factors.21 This idea is supported by case ID 7, where the staining profile suggests classical pathway despite having low Factor H levels. Since this is a retrospective study, the above hypothesis could not be validated since no genetic testing involving the alternative pathway had been performed to document a mutation that could have been precipitated by a primary infection.
As a retrospective study, there are inherent limitations. The limitations include small sample size, number of glomeruli per biopsy, lack of molecular testing of the lectin pathway, and lack of confirmatory genetic testing of the alternative pathway. However, we did see distinct differences between the C4d staining among the MCD, MPGN and C3G groups, which indicates that the staining method in itself was appropriate and useful. With samples that had fewer than six glomeruli per biopsy, sampling error and misinterpretation could be confounders. However, this often mimics realistic situations where inadequate biopsy specimens are obtained and the diagnosis is rendered on relatively small number of glomeruli. Due to tissue sample quantity, additional sections for further complement components involving the lectin pathway could not be performed. We also understand that the study has a small mixture of heterogeneous patients (both primary and secondary MPGN). However, whether the disease process is primary or secondary, the utility of C4d stain in suggesting a specific complement pathway activation is still of benefit, especially when it is interpreted as part of the complement profile composite.
In summary, MPGN and C3G are complex disease processes and further research is necessary to unravel their pathogenesis. Due to the novelty of C3G diagnosis, our center has only recently started performing a more comprehensive genetic work-up on selected patients. Investigations for factor deficiency, factor auto-antibodies, and genetic complement mutations are needed but are costly. Undoubtedly, mass spectrometry-based proteomic approaches may also be helpful, but limited in access.33 Thus, we believe that this relatively simple and widely available C4d staining, when combined with other complement stains (C1q and C3), could provide better understanding of MPGN pathogenesis and facilitate identification of C3G more easily.
Authors’ declarationsAll authors participated in the designing, conducting, or analyzing and interpreting the findings of the paper. All authors participated in writing and in reviewing its intellectual content. All authors approve the final draft of the paper attached to this declaration and approve sending it for publication in Nefrologia.
Importance of articleMPGN is a histopathological diagnosis that denotes a pattern of glomerular injury seen on light microscopy. In the past, MPGN was sub-classified into type I, II and III by immunofluorescence and electron microscopy. The first C3 Glomerulopathy consensus published in 2013 attempted to classify MPGN based on the pathogenesis involving the complement cascade. MPGN type I is believed to be due to immune complex mediated glomerular injury whereas C3G is believed to be due to the dysregulation of the alternative pathway. In this study, we define the complement cascade involvement by using the glomerular C4d and C1q profiles. These results indicate that the pathogenesis in MPGN and C3G is more complex than previously thought, and that the pathogenesis proposed by C3G consensus may warrant further investigation. In addition, for the first time, it is suggested that the lectin pathway may also be involved in the pathogenesis of MPGN in some patients.
Conflicts of interestThe authors have declared that no conflict of interest exists.



