






La glomerulopatía por invaginación podocítica (GIP) es una enfermedad de origen incierto, frecuentemente asociada a enfermedades autoinmunes, de la que se desconoce el tratamiento específico y su evolución.
Caracterizada por engrosamiento de paredes capilares por la presencia de burbujas no argirofílicas intramembanosas similares a las encontradas en la glomerulopatía membranosa, pero sin depósitos de inmunocomplejos electrodensos en la ultraestructura, donde se observan microesferas traslúcidas generadas por invaginación del citoplasma podocítico dentro de las membranas basales.
ObjetivosGeneralmente descrito en pacientes jóvenes de sexo femenino. Hasta la fecha, han sido reportados escasos casos en pacientes de origen asiático. Nuestro caso constituiría el primer reporte en paciente latinoamericano de raza blanca.
MétodosMujer de 38 años con LES. En el año 2014 presentó síndrome nefrótico tratado empíricamente con corticoides (CO) y ciclofosfamida intravenosa (CF) con buena respuesta. Presenta recaída en abril del 2015 con función renal normal y sin actividad lúpica extrarrenal, por lo que es derivada a nuestro hospital para ser biopsiada.
ResultadosLa biopsia informó esclerosis glomerular focal y segmentaria sin depósitos de inmunocomplejos en la inmunofluorescencia, pero con técnica de metenamina plata se detectaron en las paredes capilares, espacios claros acompañados de marcadas alteraciones podocíticas. Al microscopio electrónico, se observaron agregados de ultraestructuras microvesiculares y cilíndricas unidas a las membranas sin evidencia de depósitos densos y borramiento difuso de pies pedicelares, confirmando el diagnóstico sospechado.
ConclusionesReportamos el primer caso de lo que puede ser considerada, una nueva entidad patológica glomerular, en una paciente de raza blanca latinoamericana, cuya evolución y terapéutica aún se desconocen.
Podocyte infolding glomerulopathy (PIG) is a condition of uncertain origin, frequently associated with autoimmune diseases. Its specific treatment and clinical course are unknown.
It is characterised by thickening of the capillary walls due to the presence of non-argyrophilic intramembranous bubbles similar to those found in membranous glomerulopathy, but without electron-dense deposits of immune complexes in the ultrastructure, where translucent microspheres generated by invagination of the podocyte cytoplasm into the basement membranes are observed.
ObjectivesGenerally reported in young females patients. To date, few cases in Asian patients have been reported. Our case is the first to be reported in a Latin American Caucasian patient.
MethodsA 38-year-old woman with SLE. In 2014 she presented with nephrotic syndrome empirically treated with corticosteroids (CO) and intravenous cyclophosphamide with good response. She had a relapse in April 2015 with normal renal function and no extrarenal lupus activity, so she was referred to our hospital to be biopsied.
ResultsThe biopsy reported focal segmental glomerular sclerosis without deposits of immune complexes in the immunofluorescence. However, methenamine silver staining revealed clear spaces in the capillary walls accompanied by marked podocyte alterations. On electron microscope study, numerous aggregates of microvesicular and cylindrical ultrastructures bound to the membranes were observed, without evidence of dense deposits, and diffuse effacement of pedicel foot processes, confirming the suspected diagnosis.
ConclusionsThis is the first reported case of what can be considered a new pathological glomerular entity in a Latin American Caucasian patient, whose clinical course and therapy are still unknown.
La glomerulopatía por invaginación podocítica (GIP) (podocytic infolding glomerulopathy) es una enfermedad de origen incierto, frecuentemente asociada a enfermedades autoinmunes, de la que se desconoce el tratamiento específico y su evolución. Hasta la fecha han sido reportados pocos casos únicamente en pacientes de origen asiático.
Generalmente, se describe en pacientes jóvenes, con mayor prevalencia en el sexo femenino.
Se caracteriza por presentar engrosamiento de paredes capilares determinado por la presencia de pequeñas burbujas no argirofílicas intramembranosas similares a las encontradas en la glomerulopatía membranosa, pero sin depósitos de inmunocomplejos electrodensos en la ultraestructura, donde se observan en cambio microesferas traslúcidas generadas por invaginación del citoplasma podocítico dentro de las membranas basales.
Nuestro caso constituiría el primer reporte en un paciente latinoamericano y de raza blanca.
Exposición del casoMujer de 38 años de edad con antecedente de LES de 10 años de evolución. En el 2014 presentó síndrome nefrótico con función renal conservada y sedimento urinario no activo, siendo tratada con corticoides por vía oral y 6 pulsos de ciclofosfamida mensuales por vía intravenosa, con buena respuesta.
En abril del 2015 recayó el síndrome nefrótico, con función renal normal y ausencia de actividad lúpica extrarrenal, por lo que finalmente es derivada al Hospital Juan A. Fernández para realización de una biopsia renal y eventual tratamiento de la paciente.
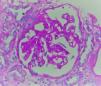
La biopsia realizada incluyó 24 glomérulos, 2globalmente esclerosados y 15 con esclerosis segmentaria. Los restantes mostraban expansión de la matriz mesangial y engrosamiento irregular de paredes capilares (fig. 1), en las que, con la técnica de metenamina plata, se reconocían burbujas no argirofílicas intramembranosas asociadas a marcadas alteraciones podocíticas que incluían hipertrofia, vacuolización, binucleación y desprendimiento (figs. 2 y 3).
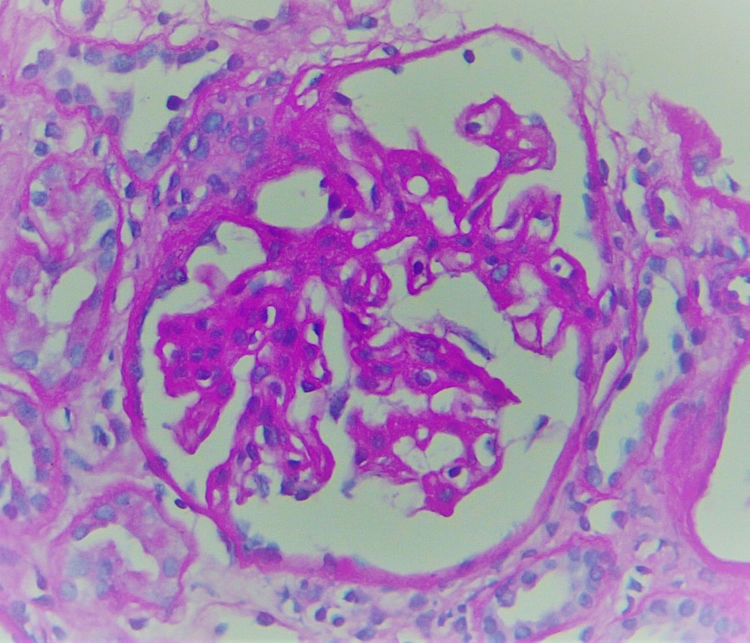
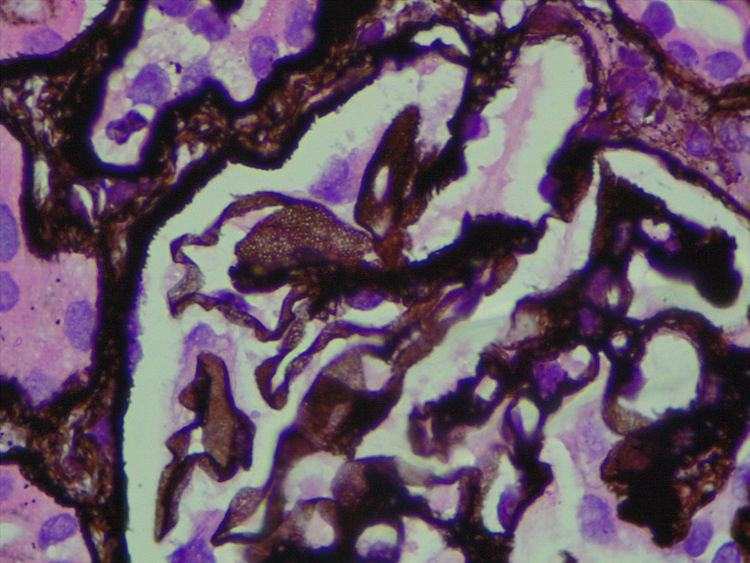
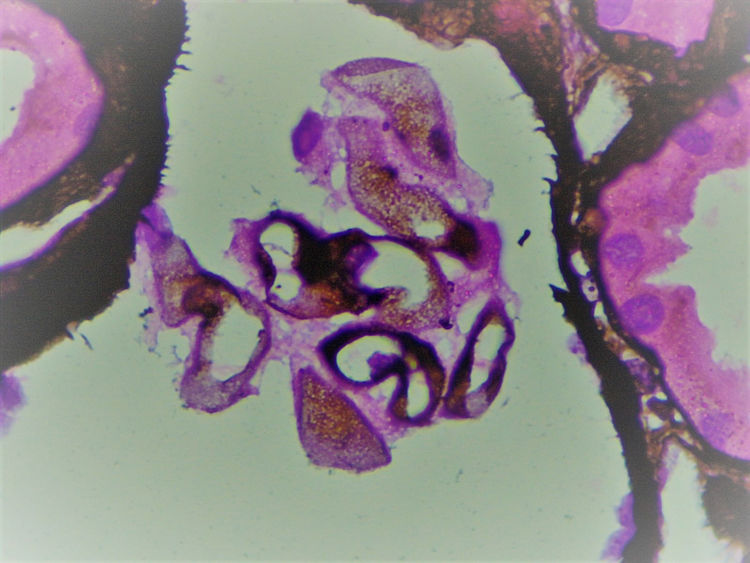
Dado que la inmunofluorescencia no demostró presencia de inmunocomplejos, resultó imprescindible realizar microscopia electrónica a fin de determinar la naturaleza de los espacios claros intraparietales.
Frente a la sospecha de una GIP, se estudió la muestra por microscopia electrónica, donde se demostró la presencia de numerosos agregados de ultraestructuras microvesiculares y cilíndricas unidas a las membranas, sin evidencia de depósitos densos y borramiento difuso de pies pedicelares (figs. 4 y 5), confirmando el diagnóstico sospechado.
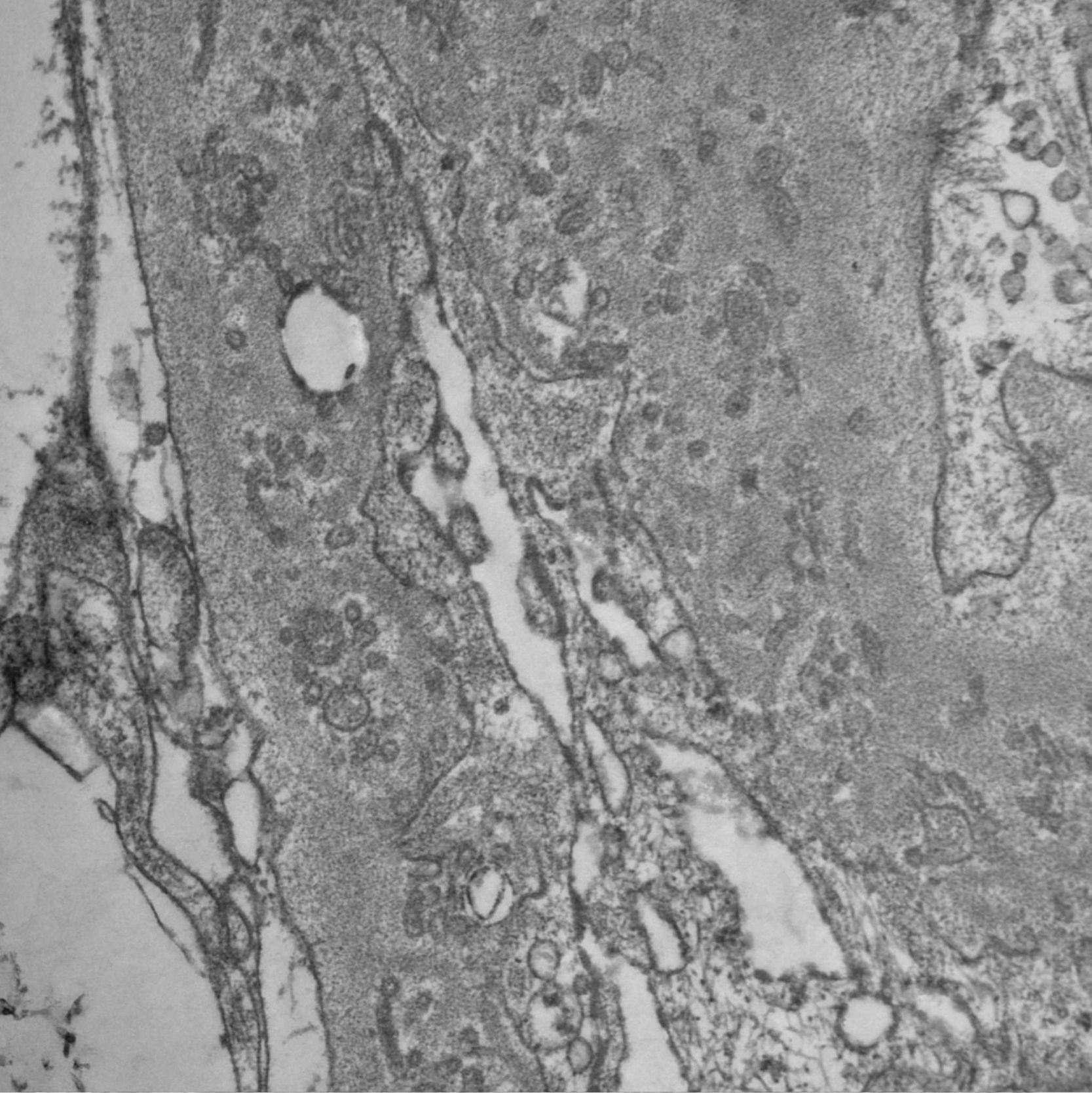
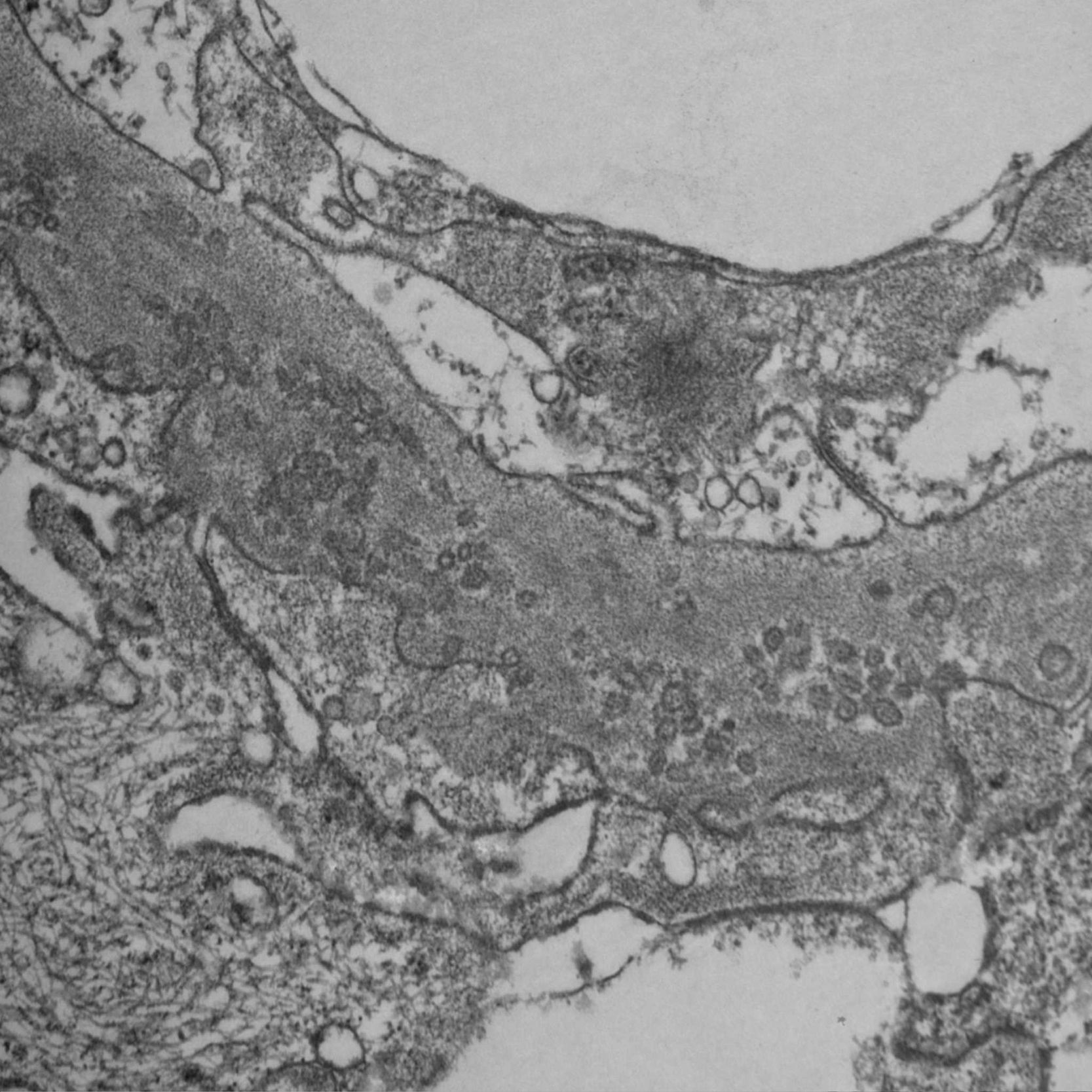
Los hallazgos histopatológicos encontrados en esta paciente son característicos de una entidad inicialmente descripta en el año 1985 como «complejo nuclear poroso» de la membrana basal glomerular (MBG)1,2 y que recientemente ha sido renombrado como GIP.
La serie más grande publicada por Joh et al.3-6, con 25 casos en Japón en el 2008, evidenció que 17 de estos pacientes presentaban enfermedades del colágeno asociadas como LES7,8 y síndrome de Sjögren9. También existen descriptos casos aislados de GIP en pacientes con mieloma múltiple10, cirrosis biliar primaria, síndrome de lisis tumoral11, arteritis de Takayasu12, hepatitis B13, reflujo vesicoureteral14 e incluso asociada a infecciones por anaerobios4.
También se asocia a patrones de glomerulopatía membranosa15,16 y esclerosis focal y segmentaria17,18.
Esto podría indicar que la GIP es consecuencia de una injuria podocitaria inespecífica19.
La GIP ha sido dividida en 2clases según predominara la «invaginación primaria» o las «microestructuras en la MBG ». Existe otra clasificación que la divide en 3 categorías, resultando de la combinación de las 2 formas anteriormente descriptas: tipo A, con invaginación podocítica primaria solamente; tipo B, con microestructuras en la MBG más invaginación podocítica primaria, y tipo C, solo con microestructuras en la MBG5.
Los criterios para definir la GIP serían: presencia de espacios no argirofílicos en la MBG detectados por microscopia óptica con técnica de metenamina plata y microesferas o microtúbulos de 50-150nm dentro de la MBG por ME20.
En la inmunofluorescencia, puede observarse tinción muy débil o negativa para IgG a lo largo de las paredes capilares.
Se ha mencionado que la invaginación del citoplasma podocitario a la MBG puede ser un patrón de alteración MBG/podocito y no una verdadera entidad patológica, ya que podría normalizarse con el cese del estímulo que lo genere. Sin embargo, la distribución extensa y severa de la lesión en la GIP hace sospechar que existen defectos intrínsecos en los mecanismos reparatorios20. El papel podocitario en la síntesis, la degradación y el mantenimiento de la MBG ha sido bien estudiado, por lo tanto, una lesión o condición anómala en el podocito o alteraciones en la producción y la degradación de la MBG podrían conducir al invaginamiento podocitario21.
Las enfermedades autoinmunes inducen fallas en la biosíntesis o en la eliminación de los componentes de la MBG por parte de los podocitos. Asimismo, el colágeno tipo iv es degradado por las metaloproteinasas, mientras que la inhibición de su degradación es producida por los inhibidores de las metaloproteinasas. De esta manera, se cree que algunos autoanticuerpos afectarían la expresión de las metaloproteinasas, estimulando así la invaginación podocitaria en pacientes con LES22.
También se ha descripto este proceso patológico asociado a enfermedades con depósitos de inmunocomplejos predominantemente con un patrón de lesión membranoso; otros pacientes muestran solamente depósitos mesangiales aislados y ocasionales subendoteliales.
Los podocitos frente a la presencia de depósitos de complejos inmunes electrodensos pueden reaccionar introduciendo su citoplasma en la MBG en el área de depósito.
La invaginación podocítica asociada a depósitos subepiteliales es común de encontrar en la nefropatía membranosa, sobre todo en estadios ii-iii.
Mediante estudios preliminares en algunos pacientes con esta condición, se llegó a indicar la presencia de un anticuerpo circulante que reaccionaría con un antígeno específico expresado por el podocito, cuya identidad precisa aún no ha podido ser determinada, pero sería diferente del receptor tipo M de la fosfolipasa A2 secretora (PLA2R) y de la trombospondina tipo 1 con el dominio 7A (THSD7A).
En algunas glomerulopatías asociadas a infecciones, la injuria glomerular puede expresarse por la presencia de partículas microesféricas en la MBG, pero en estos casos tienen una distribución focal y segmentaria, mientras en GIP las microesferas son globales y difusas23, por lo que se recomienda excluir como criterio diagnóstico de GIP la presencia de la invaginación podocítica focal y enfocarse en la presencia de microesferas y microtúbulos en la membrana basal de forma difusa24.
También se ha postulado al complejo de ataque de membrana (C5-b9) como activador podocitario, con el consecuente alargamiento de sus procesos celulares hacia el interior de la MBG alterada, pudiendo de esta manera explicar la presencia de las microestructuras intramembranosas, provenientes de la injuria podocitaria y de la MBG, causadas a su vez por inmunorreactantes del C5-b914.
Desde el punto de vista clínico, esta entidad se presenta en pacientes jóvenes, con mayor prevalencia en el sexo femenino, y suele estar asociada con proteinuria o síndrome nefrótico, habitualmente con buena respuesta a la corticoterapia.
Sin el estudio ultraestructural, esta condición es a menudo confundida y mal diagnosticada como nefropatía membranosa.
Si bien la mayoría de los pacientes presentan función renal normal, en nuestro caso, la paciente presentó falla renal debido a la mala adherencia al tratamiento inmunosupresor, y empeorado por la sepsis finalmente.
ConclusiónReportamos el primer caso de GIP en Latinoamérica y en una paciente de raza blanca. Se trata de una enfermedad de origen incierto, frecuentemente asociada a enfermedades autoinmunes. Dado que existen pocos casos publicados, aún no existen terapias específicas y se desconoce su evolución.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Helmut G. Rennke, M.D. Renal Pathology Service, Brigham y Women's Hospital, Pathology Department.



